Date: Thu, 31 Mar 2016 08:43:23 +0530
Hi,
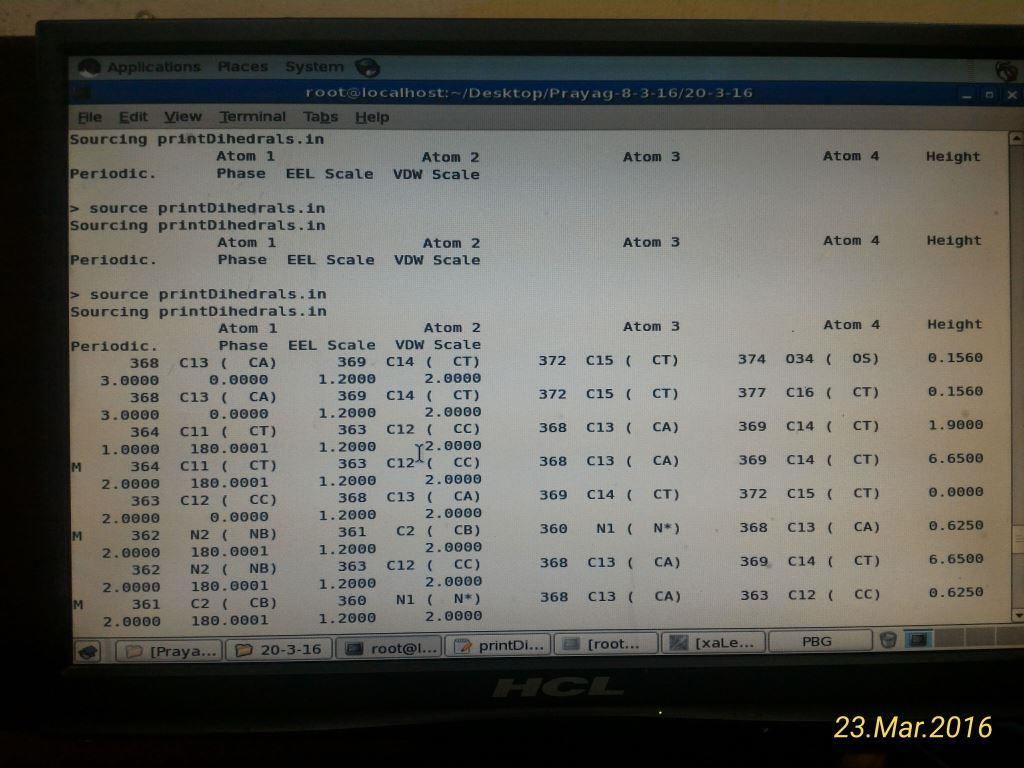
I am new to Parmed and i have tried the printdihedrals from parmed...
But i am not sure what torsions to check for??
I am experiencing trouble in each residue's glycosyl and side chain
nitrogens...
When i put those nitrogens for printdihedrals, i get a huge list of
torsions which can not be interpreted by me..
Single residue and atom selection in printdihedrals gives a smaller output
but the values given do not seem to be the torsion angle values....
I am not getting what to do with these values???
Sending herewith the frcmod file and a snapshot of parmed output..
Kindly help me.
Thanks in advance!!!
Prayagraj Fandilolu
Research Scholar,
Structural Bioinformatics Unit
Department of Biochemistry
Shivaji University, Kolhapur - 416004
(MS) India
On Tue, Mar 22, 2016 at 5:00 PM, Carlos Simmerling <
carlos.simmerling.gmail.com> wrote:
> I'm not sure if you saw the last reply by Dave Case on 3/2 - he gave you
> some useful advice but it doesn't look like you have tried that yet.
>
>
> On Tue, Mar 22, 2016 at 1:28 AM, Prayagraj Fandilolu <fprayagraj.gmail.com
> >
> wrote:
>
> > Hi David Sir,
> >
> > I took much time to reply to this mail. I did a cross check again for my
> > inputs. I reconstructed the structure using SYBYL and Chimera softwares.
> I
> > tested and derived the parameters once again.
> >
> > As you have said, i prepared a test case in which i used an RNA duplex
> > system in ff14SB. This test case is working fine no disposition form the
> > structure was observed..
> >
> > But when i am using my systems i am getting the same problem again.
> >
> > I am using xleap in which my sequence of commands is like this:
> >
> > *Using ff14SB*
> > source leaprc.ff14SB
> > source leaprc.modrna08
> > loadamberparams frcmod.ionsjc_tip3p
> > loadamberprep PMF.prepin
> > loadamberparams PMF.frcmod
> > set PMF head PMF.1.P
> > set PMF tail PMF.1.O3'
> > asl=loadpdb asl.pdb
> > check asl
> > addions asl Na+ 0
> > solvatebox asl TIP3PBOX 10
> > saveamberparm asl asl.prmtop asl.inpcrd
> >
> > *Using older ff:*
> > source leaprc.rna.ff99
> > source leaprc.ff99bsc0
> > source leaprc.modrna08
> > loadamberparams frcmod.ol.dat
> > loadamberprep PMF.prepin
> > loadamberparams PMF.frcmod
> > set PMF head PMF.1.P
> > set PMF tail PMF.1.O3'
> > asl=loadpdb asl.pdb
> > check asl
> > addions asl Na+ 0
> > solvatebox asl TIP3PBOX 10
> > saveamberparm asl asl.prmtop asl.inpcrd
> >
> > The leap log for both the cases absolutely show no error. But the
> planarity
> > is hampered in both cases. How can i check whether the prepin file is
> > faulty or not? I have previously sent all the mentioned files.
> >
> > Kindly guide me in this issue......
> >
> > Thanks in advance..
> >
> > Prayagraj Fandilolu
> > Research Scholar,
> > Structural Bioinformatics Unit
> > Department of Biochemistry
> > Shivaji University, Kolhapur - 416004
> > (MS) India
> >
> > On Wed, Mar 2, 2016 at 6:24 PM, David A Case <david.case.rutgers.edu>
> > wrote:
> >
> > > On Wed, Mar 02, 2016, Prayagraj Fandilolu wrote:
> > > >
> > > >
> > > > As you've said, I took a careful look into the leap outputs and tried
> > to
> > > > correct them with my original system.
> > > >
> > > > Today i tried loading two systems successively in two sessions.
> > > >
> > > > The warnings about improper torsion (old.log) were treated by adding
> an
> > > > additional improper into the prepin file (new.log).
> > >
> > > I think what Carlos (and others reading this) want is simpler example:
> do
> > > you have a case (preferably with a *short* oligonucleotide) where you
> get
> > > non-planar nitrogens, and that involves only standard nucleotides and
> no
> > > additional changes or parameter files? We would need the leap.in file
> > > that you used. (A good test case might involve just a single base.)
> > >
> > > The point is to find out if there is really a bug in ff14SB, or whether
> > > the problems you report are arising from changes you are making, either
> > > with modified nucleotides or with making changes to a prepin file.
> > >
> > > [Please note: on successive runs, tleap keeps adding to the end of the
> > > leap.log file, so that you can get a file that contains information
> > > about many separate tleap invocations. Please delete (or rename) your
> > > "leap.log" file before running a test case. I find it almost
> impossible
> > > to understand the files you sent for this reason, or even to locate
> what
> > > you describe as "warnings about improper torsions". And the leap.in
> file
> > > is
> > > generally more useful than the leap.log file, since the former makes it
> > > easier
> > > to see what is being done, and to try to reproduce the problem.]
> > >
> > > [Second note: if you can make a small test case, then the
> > "printDihedrals"
> > > command in parmed can be used to examine the torsion parameters in a
> > given
> > > prmtop file without having you be overwhelmed with output. Or, even
> in a
> > > bigger system, use "printDihedrals <mask>" can be used to focus
> attention
> > > on a
> > > single base.]
> > >
> > > ...thanks...dac
> > >
> > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> > >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: parmed.jpg)
- application/octet-stream attachment: mol21.frcmod
