Date: Thu, 3 Mar 2016 16:09:42 +0800 (CST)
Hello,
I calculated some complexes. Then I want to perform the MM-GB/SA calculation. When I perform "./gbsa.sh" order, it turns up the same error (shown in mmgbsa_error.jpg) for all my complexes. I don't know how to solve this problem. Please help me. Thank you very much.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
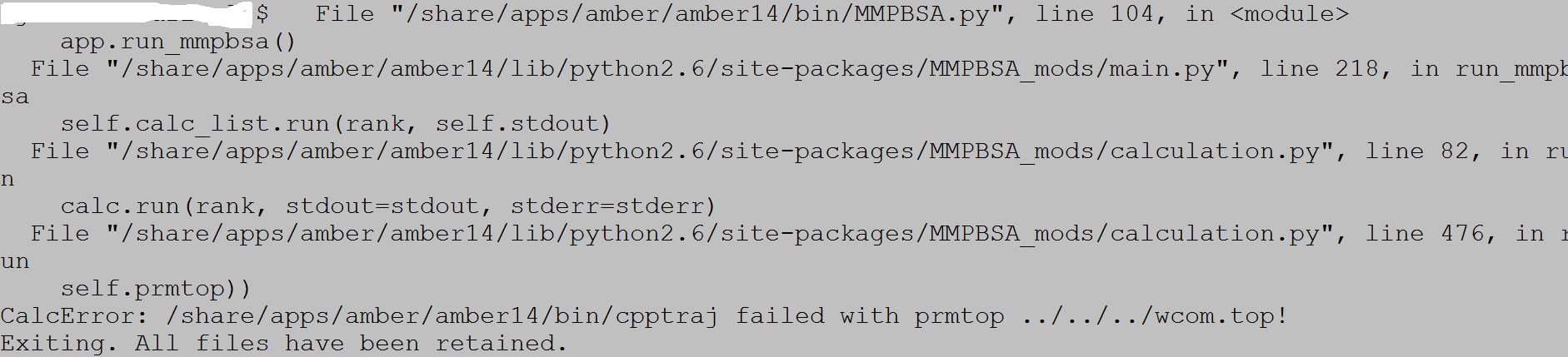
(image/jpeg attachment: mmgbsa_error.jpg)
- application/x-sh attachment: gbsa.sh
- application/octet-stream attachment: mmpbsa.in
