Date: Tue, 1 Mar 2016 17:03:44 +0530
Hello everyone,
I am doing simulation of RNA (23 nucleotides) in amber. I have followed the
tutorial given in this website(
http://ambermd.org/tutorials/basic/tutorial1/section2.htm). While while
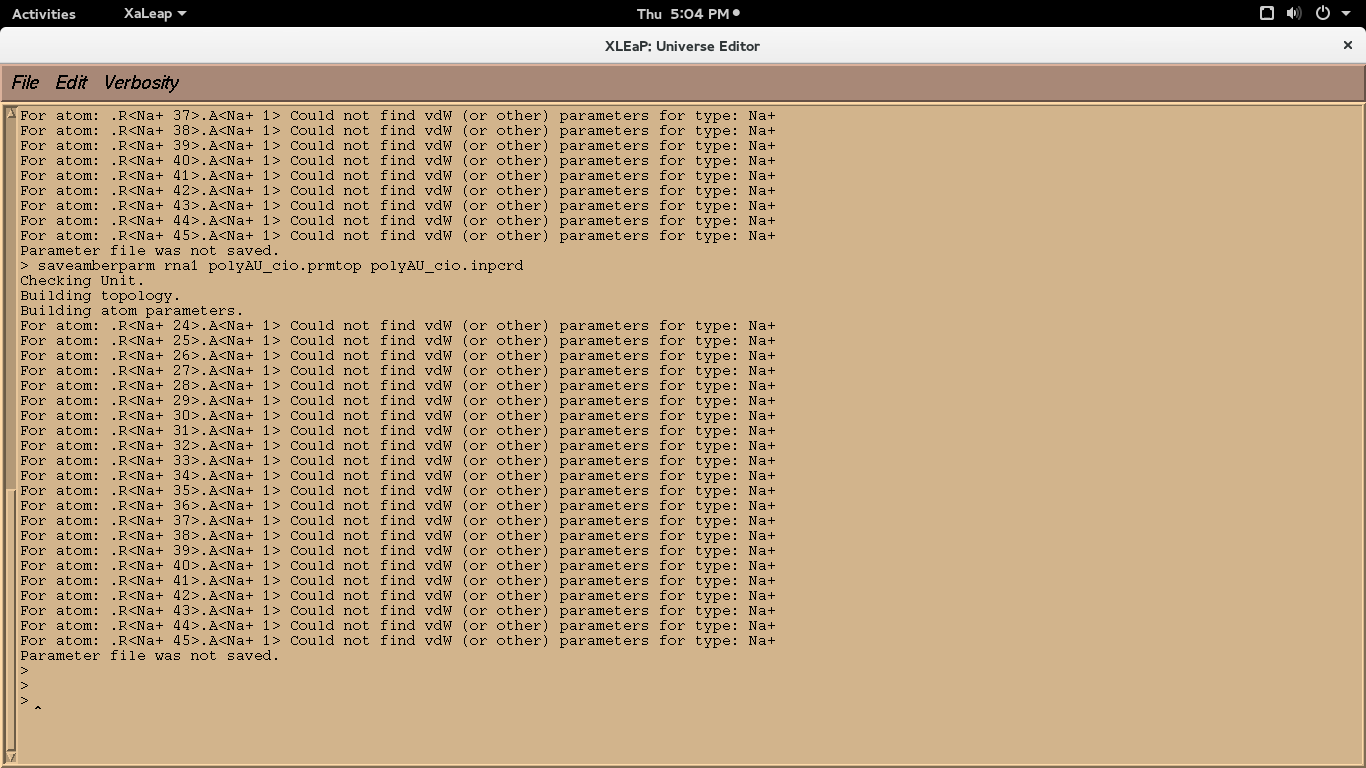
trying to write prmtop and inpcrd files for neutralised system I
encountered the following error I have pasted as snapshot. can anyone
please tell me how to rectify it??
-- With kind regards, G. Sreemol
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2016-02-25_17-04-28.png)
