Date: Mon, 30 Nov 2015 18:00:21 +0100
Dear AMBER
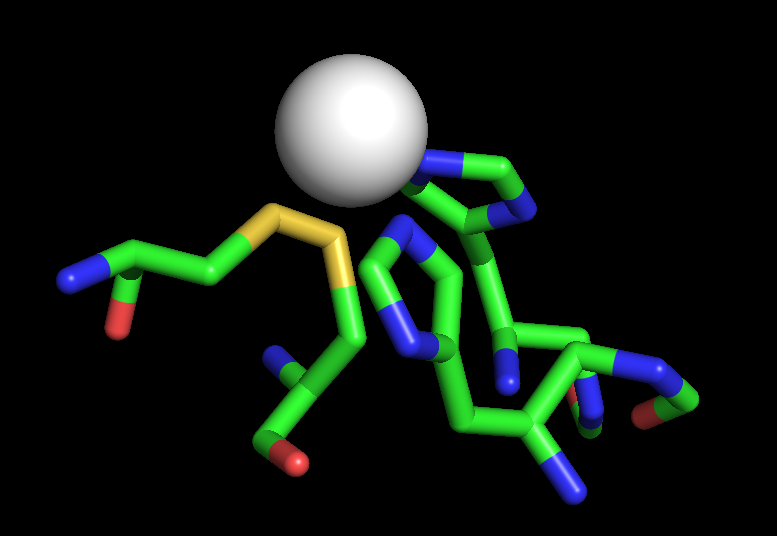
I have successfully complete refinement of my peptide with NMR restraints
on. For zinc coordination I have used following parameter (bellow). In the
end of calculation I lost perfe tetrahedral symmetry. Two cyestein residues
are forming covalent bond with each other in water or C65--SH<===>SH(C68)
about 1.5A (I have attached the image). Could any body suggest me why I
couldn't maintain proper tetrahedral geometry and why cysteins forming
covalent bonds. I am using non bonded zinc model.
thanks you in advance...
Vince
# modifications to force field for zinc finger domain
my zinc coordination frcmod file...
MASS
ZN 65.36
BOND
NB-ZN 70.000 2.05000 #kludge by JRS
ZN-S 70.000 2.10000 #kludge by JRS
ZN-SH 70.000 2.90000 #for pcy
CT-SH 222.000 1.81000 #met(aa)
ANGLE
ZN-NB-CV 50.000 126.700 #JRS estimate
ZN-NB-CR 50.000 126.700 #JRS estimate
ZN-NB-CP 50.000 126.700 #JRS estimate
ZN-NB-CC 50.000 126.700 #JRS estimate
ZN-SH-CT 50.000 120.000 #JRS estimate
ZN-S -CT 50.000 120.000 #JRS estimate
ZN-S -C2 50.000 120.000 #JRS estimate
ZN-S -C3 50.000 120.000 #JRS estimate
NB-ZN-NB 10.000 110.000 #dac estimate
NB-ZN-SH 10.000 110.000 #dac estimate
NB-ZN-S 10.000 110.000 #dac estimate
SH-ZN-S 10.000 110.000 #dac estimate
ZN-SH-CT 50.000 120.000 #JRS estimate
CT-CT-SH 50.000 114.700 #met(aa)
HC-CT-SH 35.000 109.500
H1-CT-SH 35.000 109.500
CT-SH-CT 62.000 98.900 #MET(OL)
SH-ZN-SH 10.00 113.000 #by VK
S-ZN-S 10.00 109.100 #by VK
DIHE
X -NB-ZN-X 1 0.000 180.000 3.000
X -ZN-SH-X 1 0.000 180.000 3.000
X -ZN-S -X 1 0.000 180.000 3.000
X -CT-SH-X 3 1.000 0.000 3.000
NONBON
ZN 1.10 0.0125
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: zinc_cordination.png)
