Date: Mon, 9 Nov 2015 15:05:31 +0000
Many thanks for your valuable advice; I have been playing with different
values for the reaction coordinate to improve the overlap and what I am
using now is a difference ccordinate between the bonds to be formed and the
one to be broken; at the beginning the windows are 0.1 A with a force
constant of 100 and then the seperation becomes 0.05 A near the transition
state with a force constant of 300. The overlap is very good with these
values. Later on I use the WHAM program from Alan Grossfield to unbiase the
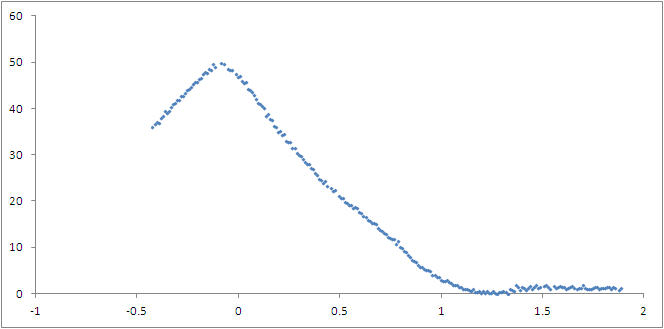
potential. The result is in the next graph:
[image: Inline images 1]
The PMF of the products (on the left side of the graph) is much higher than
that of the reactants. Is that OK? How would I calculate the activation
energy for the reaction?
Many thanks,
Mahmood
On 27 October 2015 at 12:30, Adrian Roitberg <roitberg.ufl.edu> wrote:
> Hi
>
> When we do this, for a simple bond breaking/forming, we tend to use
> windows roughly 0.1 apart, and force constants of the order of 200.
>
> See like the overlap is better than before, but I would have more
> windows between the ones you have now. Try also much larger force
> constant around your transition state, which is probably around a value
> of zero for your generalized coordinate.
>
> Adrian
>
>
> On 10/27/15 8:05 AM, Mahmood Jasim wrote:
> > Hi Adrian,
> >
> > Many thanks for your comments; they were very useful. Regarding ig==-1, I
> > am using it except in the single input file I copied in the email. I am
> now
> > running the simulation using the generalized distance coordinate as the
> > difference between the lenghths of the bond to be formed and the one to
> be
> > broken with 0.2 A windows. This is my restraint file:
> >
> > iat=1937,6803,6803,6798, r1=-10.0, r2=1.5, r3=1.5, r4=10.0, rstwt=1,-1,
> > rk2=50, rk3=50,
> >
> > and the overlap has greatly improved. The problem starts to happen when
> the
> > coordinate gets closer to the formation of the bond where a big gap
> appears
> > in the graph and the coordinate jumps from 0.15 to -0.3 A. I tried
> > increasing the force near the region but it did not work. This is how the
> > reaction coordinate versus time looks like:
> >
> > [image: Inline images 2]
> >
> > Any advice please
> >
> > Best Regards,
> > Mahmood
> >
> > On 23 October 2015 at 14:39, Adrian Roitberg <roitberg.ufl.edu> wrote:
> >
> >> Hi
> >>
> >> Several comments.
> >>
> >> Never run the WHOLE set of simulations before you are satisfied with the
> >> overlap. Just run 3 or 4 windows and then see if you need to change
> >> something before going further.
> >>
> >> As for your qeustions:
> >>
> >> Is that sufficient overlap? If it is not, do I need to reduce the
> spacing
> >> or increase the force constant to get better overlap?
> >>
> >> You do not show the overlap, so it is hard to tell you, but my guess
> >> from your first figure is that no, it is not enough. By the way, if you
> >> want to increase overlap, you need either a closer spacing and/or a
> >> LOWER force constant(not increased!).
> >>
> >> PLEASE add ig=-1 to your mdin !
> >>
> >> As for your coordinate, it is NEVER a good idea to use just one distance
> >> for a reaction. Look at published work on this, and you will see that
> >> peopel tend to use a reaction coordinate that has RC=distance(bond being
> >> broken) - distance(bond being made)
> >>
> >> That produced much more stable surfaces.
> >>
> >> adrian
> >>
> >>
> >>
> >> On 10/23/15 7:56 AM, Mahmood Jasim wrote:
> >>> Hi AMBER users,
> >>>
> >>> I am trying to simulate a reaction involving the formation of a
> covalent
> >>> bond using umbrella sampling combine with QM treatment of the reaction
> >>> atoms. I have the SG atom of a cysteine residue attacking an
> electrophilc
> >>> carbon on a ligand. The distance between these 2 atoms is the reaction
> >>> coordinate. The windows of umbrella sampling are spaced 0.3 Angstrom
> >> apart.
> >>> The restraint file looks like this:
> >>>
> >>> #
> >>> # 123 CYM SG 432 UNK C1 3.43
> >>> &rst
> >>> ixpk= 0, nxpk= 0, iat=1937,6803, r1= 2.93, r2= 3.43, r3= 3.43, r4=
> >> 3.93,
> >>> rk2=0.0, rk3=100.0, ir6=1, ialtd=0,
> >>> &end
> >>>
> >>> and the md.in file looks like this:
> >>>
> >>> &cntrl
> >>> imin = 0,
> >>> irest = 1,
> >>> ntx = 7,
> >>> ntb = 2,
> >>> cut = 12,
> >>> ntr = 0,
> >>> ntc = 1,
> >>> ntf = 1,
> >>> igb = 0
> >>> ntp = 1
> >>> tempi = 300.0,
> >>> temp0 = 300.0,
> >>> ntt = 3,
> >>> gamma_ln = 1.0,
> >>> nstlim =10000, dt = 0.001
> >>> ntpr = 1000, ntwx = 1000, ntwr = 1000, nmropt = 1,
> >>> ifqnt=1,
> >>> /
> >>>
> >>>
> >>> &qmmm
> >>>
> >>> qmmask =
> >>> '.1934-1937,6794-6848',
> >>> qmcharge =
> >>> 0,
> >>> qm_theory =
> >>> 'PM3',
> >>> qmcut =
> >>> 12.0,
> >>> /
> >>> &wt type='DUMPFREQ', istep1=100,/
> >>> &wt type='END' /
> >>> DISANG=RST-1.dist
> >>> LISTOUT=POUT
> >>> DUMPAVE=DIST_1.dat
> >>>
> >>> The run was able to simulate the formation of the bond at the end and
> the
> >>> next graph represents the values of the coordinates across the windows.
> >>> [image: Inline images 1]
> >>>
> >>> Is that sufficient overlap? If it is not, do I need to reduce the
> spacing
> >>> or increase the force constant to get better overlap?
> >>>
> >>> I used the WHAM code by Alan Grossfieldto calculate PMF with this
> >> command:
> >>> wham 1.59 3.73 70 0.00001 300 0 meta.dat result.dat
> >>>
> >>> and this was the meta.dat file:
> >>>
> >>> DIST_1.dat 3.43 200
> >>> DIST_2.dat 3.13 200
> >>> DIST_3.dat 2.83 200
> >>> DIST_4.dat 2.53 200
> >>> DIST_5.dat 2.23 200
> >>> DIST_5.dat 1.93 200
> >>> DIST_7.dat 1.63 200
> >>>
> >>> and the histogram I am getting looks like:
> >>>
> >>> [image: Inline images 2]
> >>>
> >>> I was expecting the histogram to look different, the PMF to go higher
> and
> >>> then decreases as it approcahes the products. If I managed to improve
> the
> >>> overlapping, will this change the histogrm?
> >>>
> >>> Many thanks,
> >>> Mahmood Jasim
> >>> Aston University
> >>> UK
> >>>
> >>>
> >>>
> >>> _______________________________________________
> >>> AMBER mailing list
> >>> AMBER.ambermd.org
> >>> http://lists.ambermd.org/mailman/listinfo/amber
> >> --
> >> Dr. Adrian E. Roitberg
> >> Professor.
> >> Department of Chemistry
> >> University of Florida
> >> roitberg.ufl.edu
> >> 352-392-6972
> >>
> >> _______________________________________________
> >> AMBER mailing list
> >> AMBER.ambermd.org
> >> http://lists.ambermd.org/mailman/listinfo/amber
> >>
> >
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
> --
> Dr. Adrian E. Roitberg
> Professor.
> Department of Chemistry
> University of Florida
> roitberg.ufl.edu
> 352-392-6972
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
