Date: Mon, 21 Sep 2015 16:55:55 -0300
Dear AMBER of the list,
I'm trying to use as the initial approximation method
a polynomial fit high school (harmonic approximation)
to obtain the force constants for the file .frcmod
However, reading the manual I found information about the function
MASKS that from what I understand one can freeze specific regions
for a DM calculation.
This works is like that .... ????
Another thing that has been tried before using antechamber and parmchk
obitive
certain force constants values (some literature) and minimum distances
coming calculations
optimization means gaussian. With this .frcmod file with a little empirical
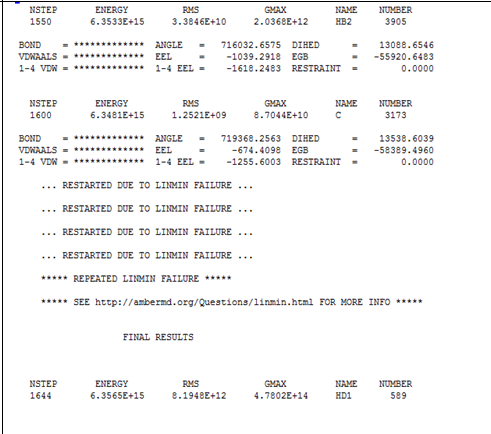
values I tried to hold a DM entering metals (two Zn) in the active site
via unbound model (free ion). But the DM minimization in one step
does not end all the steps and the error appears below. A minimization in
stages
I could fix it (for an initial test because I'm trying to parameterize the
entire active site)? How to do this???
[image: Imagem intercalada 1]
Best regards
Marcelo Andrade Chagas, MSc
(PhD student)
Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
* http://lqcmm.qui.ufmg.br/
Departamento de Química da Universidade Federal de Minas Gerais - UFMG
Tel:(31)3409-5776
2015-09-21 16:33 GMT-03:00 Gustavo Seabra <gustavo.seabra.gmail.com>:
> Hi Marcelo,
>
> You cannot run MD if there are missing parameters for your system. Just
> “freezing” some of the atoms will not work.
>
> What I’d suggest is that, if parameters are not available, you should try
> to generate the missing parameters by using one of the many available
> methods. The choice of the specific method you use may depend on your
> system. With more details, some people on this list may be able to help you
> choose.
>
> Gustavo Seabra
>
>
>
> > Em 21/09/2015, à(s) 16:21, Marcelo Andrade Chagas <
> andrade.mchagas.gmail.com> escreveu:
> >
> > Dear AMBER of the list,
> >
> > how to freeze atoms in an active site of a protein
> > that do not have parameters and perform an MD ???
> >
> > Best regards
> >
> > Marcelo
> >
> > Marcelo Andrade Chagas, MSc
> > (PhD student)
> > Laboratório de Química Computacional e Modelagem Molecular - LQC-MM
> > * http://lqcmm.qui.ufmg.br/
> > Departamento de Química da Universidade Federal de Minas Gerais - UFMG
> > Tel:(31)3409-5776
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
