On Fri, Jun 5, 2015 at 11:26 AM, Indrajit Deb <biky2004indra.gmail.com>
wrote:
> Dear amber users,
>
> I have a average pdb file which have short contacts. I want to convert the
> average pdb file into rst file, so that I can use it as an input for
> sander. Actually I want to energy minimize the average pdb.
>
Take care here -- average PDBs are very frequently non-physical. For
example, freely rotating methyl groups will have all hydrogen atoms in the
same place, almost on top of the carbon atom (since that's its "average"
position). Even minimizing the structure may prove challenging. Also look
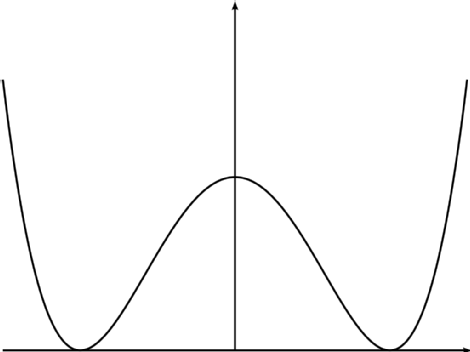
at a typical double-well potential like the following:
http://www2.warwick.ac.uk/fac/sci/masdoc/current/msc-modules/ma916/mathematics_of_multiscale_materials/purenearestneighbourinteraction/examples/doublwell3.png?maxWidth=471&maxHeight=353
Assuming that surface is fully-sampled, the "average" structure will be the
maximum energy structure between the two wells, which isn't representative
of *any* dominant configuration. And minimizing it will put you in one of
the two wells, which will no longer be anything like the average structure
(assuming you're *slightly* off the exact average).
It's not clear to me exactly what you should expect when minimizing an
average structure.
So how to convert average pdb file into rst file ?
>
cpptraj does this in a single command:
cpptraj -p average.pdb -y average.pdb -x average.rst7
HTH,
Jason
--
Jason M. Swails
BioMaPS,
Rutgers University
Postdoctoral Researcher
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Fri Jun 05 2015 - 09:00:02 PDT
{kind=link}
