Date: Mon, 27 Apr 2015 21:19:57 -0400
Good evening!
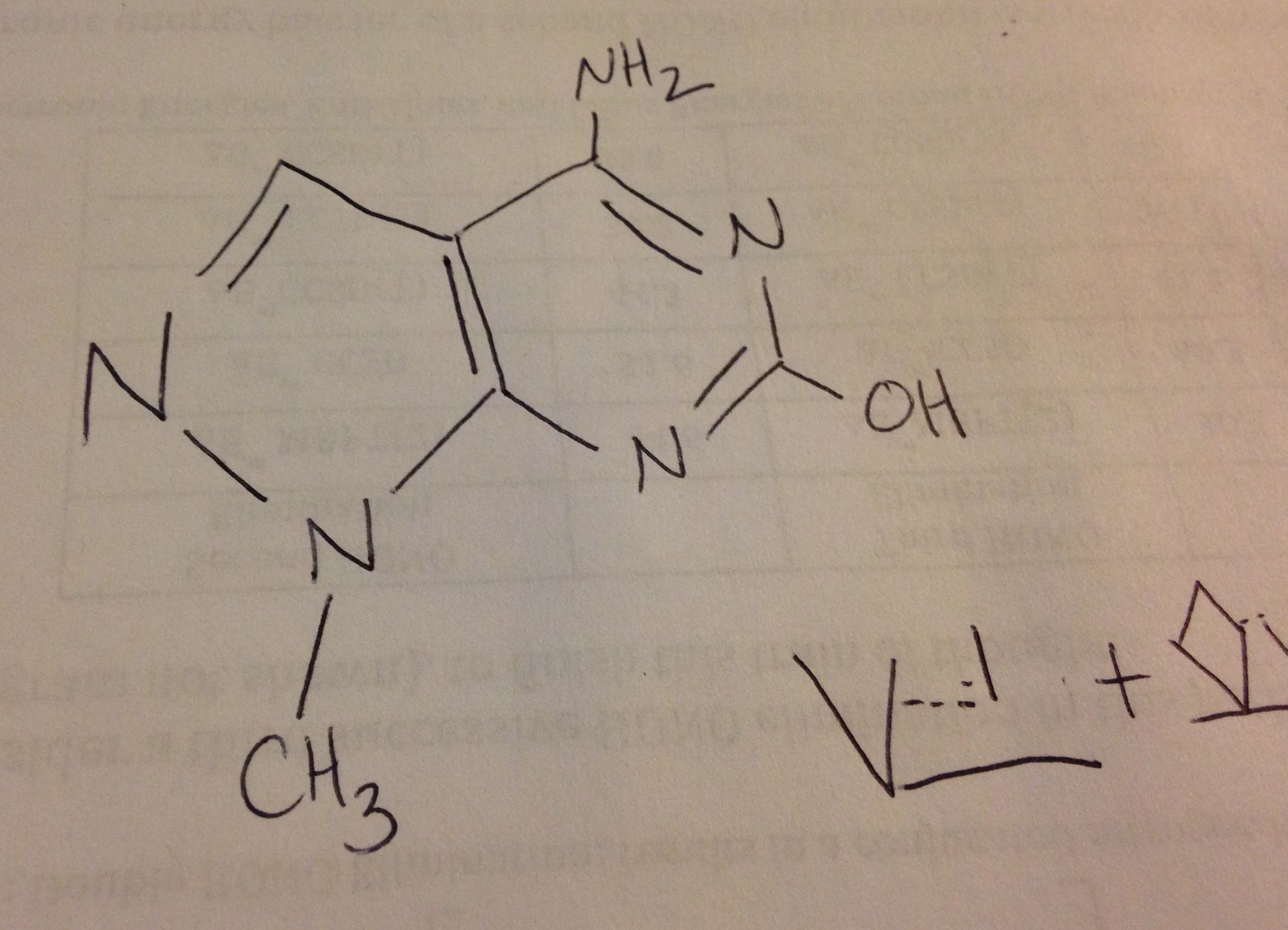
I have a small, planar, aromatic organic molecule (C, N, H, O atoms). I
encountered a problem in parameterizing with GAFF; 4 dihedrals were not
available. I tried to identify the four atoms in the dihedral in my
molecule, so I could just calculate the torsional PES and add the
parameters, and found some challenges. I have attached a photo of the
molecule in question.
a.) Specifically, I was informed that I lacked 2 ca-ca-cc-h4 dihedral
parameters and 2 ca-ca-cc-nd dihedral parameters by xleap. However, when
I look at the molecule, this designation does not seem to make sense.
There are 3 ca type carbons in the molecule, if I understand properly,
and none of them are adjacent to one another; there is no cc adjacent to
a h4. I /assume/, perhaps erroneously, that the "best" choice in
dihedrals to form will be the ones that do not "jump" over atoms and
thus go over grater distances.
b.) To confirm this, I examined ANTECHAMBER_AC.AC. It lists the
following table:
ATOM 1 C1 MOL 1 -1.428 -1.191 -0.001 0.000000 ca
ATOM 2 N1 MOL 1 -2.039 0.009 0.000 0.000000 nb
ATOM 3 C2 MOL 1 -1.280 1.097 -0.003 0.000000 ca
ATOM 4 C3 MOL 1 0.124 0.968 -0.003 0.000000 ca
ATOM 5 C4 MOL 1 0.596 -0.349 0.001 0.000000 ca
ATOM 6 N2 MOL 1 -0.146 -1.465 0.002 0.000000 nb
ATOM 7 C5 MOL 1 1.299 1.764 -0.006 0.000000 cc
ATOM 8 H1 MOL 1 1.405 2.837 -0.014 0.000000 h4
ATOM 9 N3 MOL 1 -1.900 2.297 -0.029 0.000000 nh
ATOM 10 H2 MOL 1 -2.896 2.310 0.100 0.000000 hn
ATOM 11 H3 MOL 1 -1.380 3.137 0.142 0.000000 hn
ATOM 12 N4 MOL 1 2.371 1.010 -0.003 0.000000 nd
ATOM 13 N5 MOL 1 1.942 -0.275 0.002 0.000000 na
ATOM 14 C6 MOL 1 2.878 -1.373 0.005 0.000000 c3
ATOM 15 H4 MOL 1 3.505 -1.324 0.893 0.000000 h1
ATOM 16 H5 MOL 1 2.301 -2.294 0.006 0.000000 h1
ATOM 17 H6 MOL 1 3.506 -1.327 -0.883 0.000000 h1
ATOM 18 O1 MOL 1 -2.253 -2.246 -0.001 0.000000 oh
ATOM 19 H7 MOL 1 -3.150 -1.895 -0.005 0.000000 ho
To me, this seemingly confirms my suspicion in a.) This table lists
there to be 4 ca's in the molecule and one cc. However, pursuant to the
original GAFF paper, I would say that there are 3 ca's and 2 cc's in the
molecule (by my eye). Either that or I deserve an F in organic
chemistry, and hopefully Dr. Farrell is not on this list-serv.
c.) I am wondering if the antechamber algorithm for designating atom
types is meeting a pathological case? i.e., that it is assigning
incorrect atom types? I would guess that antechamber uses a
distance-based criterion for designating atom types? I am having trouble
finding out in the manual how antechamber takes xyz coordinates read in
to decide atom types?
-- Dr. Robert Molt Jr. r.molt.chemical.physics.gmail.com Nigel Richards Research Group Department of Chemistry & Chemical Biology Indiana University-Purdue University Indianapolis LD 326 402 N. Blackford St. Indianapolis, IN 46202
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: molecule.jpg)
