Date: Fri, 6 Mar 2015 16:26:24 +0800
Dear Francois,
Thanks a lot for providing such invaluable information. I've read the
tutorial and run two jobs on the R.E.D. Develoment server. However,
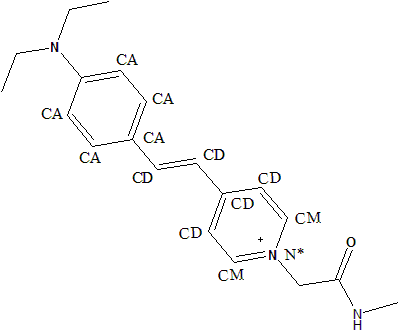
the atom typing of one looks a little strange, so I am looking for help for
this molecule (see attached picture).
Generally, this molecule was two aromatic ring linked by ethylene. The
R.E.D server could correctly calculate the partial charge for it, but the
assigned atom types surprised me. The two carbon atom from ethylele was
assigned atom type "CD", which was mentioned in parm99.dat as "sp2 C atom
in the middle of: C=CD-CD=C". But, in our case, the two carbon atoms was
connected by double bond, not single bond. I also found the description of
"CD" from Wang et al. 2000, which said that,
"Considering that the inner two carbons of butadiene are not pure sp2
carbons, and the bond lengths between them are slightly longer than a pure
single Csp2—Csp2, a new atomic type (CD) can be introduced. "
So, I am a little confused. Is CD the proper atom type for those two
carbons? Or do you have any experience/examples on such conjugated systems?
Many Thanks & Best Regards,
Tao
2015-03-03 17:31 GMT+08:00 FyD <fyd.q4md-forcefieldtools.org>:
> Dear Tao,
>
> See http://q4md-forcefieldtools.org/REDServer-Development/
> and its tutorial:
> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php
>
> and particularly:
> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php#17
> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php#18
>
> in agreement with:
> http://onlinelibrary.wiley.com/doi/10.1002/jcc.540161106/abstract
>
> regards, Francois
>
> > I am doing simulation on system that with various compounds bonded to the
> > N-terminal of certain peptide, like shown in the following picture. Now I
> > am facing the problems with creating unit for such N-Cap residue.
> >
> > ?
> > 1. Should I treat R1 combined R' as a N-Cap residue or just the R' only?
> > 2. In any case above, is there any method available to assign correct
> > partial charge and atom type for such non-standard residue?
>
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: L2T.png)
