Dear Joe,
> Thanks for the helpful reply. I tried running with Re_Fit = on, and filled
> in the missing OH-P -OH angle parameter by analogy to the O2-P -O2
> parameter in the frcmod.known file from the first run, uploading that as
> the frcmod.user file. I'm a little uncertain about what you mean by
> "without your own mandatory parameters", however.
See
http://q4md-forcefieldtools.org/REDServer-Development/faq.php#5
When describing the problem you encountered with R.E.D. Server
Development, please also provide the 'PXXXX' R.E.D. Server Development
job name in the body of your email so that we can more easily assist
you.
If I look at the P20692 (Re_Fit) job (I searched the email database
using your email address and selected your last job): you provided a
frcmod.user file using/completing the frcmod.unknown file from the
previous job; your new frcmod.unknown file is now empty; Seems pretty
good...
You can see that the energy decomposition is carried: (because there
is now no missing FF parameter)
http://cluster.q4md-forcefieldtools.org/~x/Project/P20692/Data-R.E.D.Server/Mol_m1/Energy_m1-c1.txt
http://cluster.q4md-forcefieldtools.org/~x/Project/P20692/Data-R.E.D.Server/Mol_m2/Energy_m2-c1.txt
http://cluster.q4md-forcefieldtools.org/~x/Project/P20692/Data-R.E.D.Server/Mol_m3/Energy_m3-c1.txt
-> single point ; you could compare with what you get with sander.
Important point: check the conformation(s) obtained after geometry
optimization: i.e. the Mol_m$n/Mol_m$n-c$i-qmra.pdb files: are they in
agreement with what you want?
Concerning 'mandatory' FF parameters: let's say you do not agree with
a FF parameter available in the frcmod.known or frcmod.correspondence
files; i.e. with a FF parameter automatically determined by PyRED from
the internal database of Amber FF. Just provide that unwanted FF
parameter (with the missing one) in the frcmod.user file and it will
replace that automatically determined by PyRED. Test & see ;-)
regards, Francois
> On Wed, Mar 4, 2015 at 3:56 AM, FyD <fyd.q4md-forcefieldtools.org> wrote:
>
>> Dear Joe,
>>
>> A google search at http://archive.ambermd.org/ gives:
>> http://archive.ambermd.org/201203/0076.html
>> http://archive.ambermd.org/201208/0388.html
>>
>> or just read i.e.
>> http://q4md-forcefieldtools.org/REDDB/projects/F-90/script1.ff
>>
>> perl -p -i.old -e 's/\#\#\!\# //g' script1.ff
>> grep -v "to be replaced by the correct tripos" script1.ff > script1-new.ff
>> # up to you...
>> # perl -p -i.old -e 's/\#\#\#\#\#\# //g' script1-new.ff
>>
>> & look for ATP1 ATP2 dATP1 dATP2
>>
>> Concerning hydrogen phosphate, dihydrogen phosphate, ...:
>> See http://q4md-forcefieldtools.org/REDDB/projects/F-90/script4.ff
>> -> the answer is no
>>
>> It looks like data are below ;-) generated using R.E.D. Server Dev./PyRED
>> This could give you a starting point for other derivatives:
>>
>> http://q4mdfft:q4mdfft2012.cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650.html
>>
>> The 3 FF libs for the 3 input molecules:
>>
>> http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm1-c1.mol2
>>
>> http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm2-c1.mol2
>>
>> http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm3-c1.mol2
>>
>> There is one missing FF paramter: a big deal ;-)
>>
>> http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Data-Default-Proj/frcmod.unknown
>> To be added in a sec using the 'Re_Fit' mode & a frcmod.user input
>> file (with or without your own mandatory parameters):
>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php
>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo1.pdf
>>
>> Energy decomposition is only carried out - when... there is
>> (obviously) no missing force filed parameters.
>>
>> regards, Francois
>>
>>
>> > Thanks for the links. I took a look at the RED DB info, and it is not
>> clear
>> > to me how to manipulate what is in that database to obtain an inorganic
>> > hydrogen phosphate and inorganic dihydrogen phosphate. Is that possible
>> > using what is in the F-90 project?
>>
>>
>> > On Tue, Mar 3, 2015 at 4:28 AM, FyD <fyd.q4md-forcefieldtools.org>
>> wrote:
>> >
>> >> Dear Joseph,
>> >>
>> >> See also the F-90 REDDB project:
>> >> http://q4md-forcefieldtools.org/REDDB/projects/F-90/
>> >> http://q4md-forcefieldtools.org/REDDB/projects/F-90/script1.ff
>> >>
>> >> On the top of the FF parameters you can always add your own set of FF
>> >> paramters.
>> >>
>> >> R.E.D. Server/R.E.D. IV are outdated!
>> >> Please use instead: R.E.D. Server Dev./PyRED at
>> >> http://q4md-forcefieldtools.org/REDServer-Development/
>> >>
>> >> See http://q4md-forcefieldtools.org/REDServer/images/AnteRED-RED.gif
>> >> vs
>> >> http://q4md-forcefieldtools.org/REDServer-Development/images/PyRED.gif
>> >>
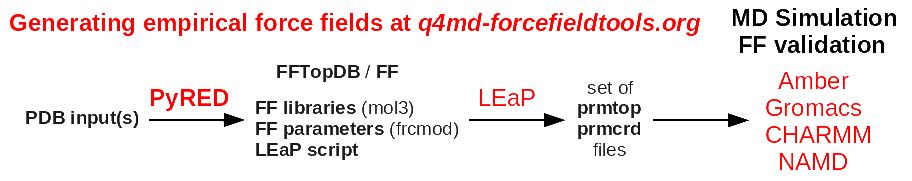
>> >> PyRED generate a whole force field i.e. not only atomic charges.
>> >>
>> >> See the tutorial:
>> >> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php
>> >> & in particular the last ones:
>> >> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo1.pdf
>> >> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo5.pdf
>> >> where you can provide your own atom types (or correct theses
>> >> automatically generated) and mandatory/missing force field parameters...
>> >>
>> >> the mol3 file format is the default file format for FF libraries:
>> >> http://q4md-forcefieldtools.org/Tutorial/leap-mol3.php
>> >> (it is advantageous for molecular fragments)
>> >>
>> >> regards, Francois
>> >>
>> >>
>> >>
>> >> > I see that the Carlson group has some ADP and ATP parameters that have
>> >> been
>> >> > developed for use with ff99SB. However, from my understanding, ff12SB
>> or
>> >> > ff14SB are now recommended for protein simulation due to some
>> >> improvements
>> >> > in how they treat protein secondary structure. Is anybody aware of
>> >> whether
>> >> > the Carlson ADP/ATP parameters have been vetted against ff12SB or
>> ff14SB,
>> >> > or if one should expect the modifications that went into those
>> >> force-fields
>> >> > to break anything badly if they are used with the Carlson ADP/ATP
>> >> > parameters? I'm curious because I saw a recent (2013) nice paper from
>> the
>> >> > Hummer group on F1-ATPase where they still used the combination of
>> ff99SB
>> >> > with these Carlson parameters.
>> >> >
>> >> > Also, does anyone know if there are any parameters for HPO42- or
>> H2PO4-
>> >> > floating around out there that have been tested with Carlson ADP +
>> >> > ff99SB/ff12SB/ff14SB? If not then I'll go with the antechamber + RED
>> >> server
>> >> > route.
>>
>>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Thu Mar 05 2015 - 00:00:02 PST
{kind=link}
{kind=link}
