Dear Joe,
A google search at
http://archive.ambermd.org/ gives:
http://archive.ambermd.org/201203/0076.html
http://archive.ambermd.org/201208/0388.html
or just read i.e.
http://q4md-forcefieldtools.org/REDDB/projects/F-90/script1.ff
perl -p -i.old -e 's/\#\#\!\# //g' script1.ff
grep -v "to be replaced by the correct tripos" script1.ff > script1-new.ff
# up to you...
# perl -p -i.old -e 's/\#\#\#\#\#\# //g' script1-new.ff
& look for ATP1 ATP2 dATP1 dATP2
Concerning hydrogen phosphate, dihydrogen phosphate, ...:
See
http://q4md-forcefieldtools.org/REDDB/projects/F-90/script4.ff
-> the answer is no
It looks like data are below ;-) generated using R.E.D. Server Dev./PyRED
This could give you a starting point for other derivatives:
http://q4mdfft:q4mdfft2012.cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650.html
The 3 FF libs for the 3 input molecules:
http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm1-c1.mol2
http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm2-c1.mol2
http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Mol_MM/Mol_mm3-c1.mol2
There is one missing FF paramter: a big deal ;-)
http://cluster.q4md-forcefieldtools.org/~ucpublic1/ADF1ADFVTxTnW3LADFIRLutnVSdsCY2OeP4Jz3Z1/P20650/Data-R.E.D.Server/Data-Default-Proj/frcmod.unknown
To be added in a sec using the 'Re_Fit' mode & a frcmod.user input
file (with or without your own mandatory parameters):
http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php
http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo1.pdf
Energy decomposition is only carried out - when... there is
(obviously) no missing force filed parameters.
regards, Francois
> Thanks for the links. I took a look at the RED DB info, and it is not clear
> to me how to manipulate what is in that database to obtain an inorganic
> hydrogen phosphate and inorganic dihydrogen phosphate. Is that possible
> using what is in the F-90 project?
> On Tue, Mar 3, 2015 at 4:28 AM, FyD <fyd.q4md-forcefieldtools.org> wrote:
>
>> Dear Joseph,
>>
>> See also the F-90 REDDB project:
>> http://q4md-forcefieldtools.org/REDDB/projects/F-90/
>> http://q4md-forcefieldtools.org/REDDB/projects/F-90/script1.ff
>>
>> On the top of the FF parameters you can always add your own set of FF
>> paramters.
>>
>> R.E.D. Server/R.E.D. IV are outdated!
>> Please use instead: R.E.D. Server Dev./PyRED at
>> http://q4md-forcefieldtools.org/REDServer-Development/
>>
>> See http://q4md-forcefieldtools.org/REDServer/images/AnteRED-RED.gif
>> vs
>> http://q4md-forcefieldtools.org/REDServer-Development/images/PyRED.gif
>>
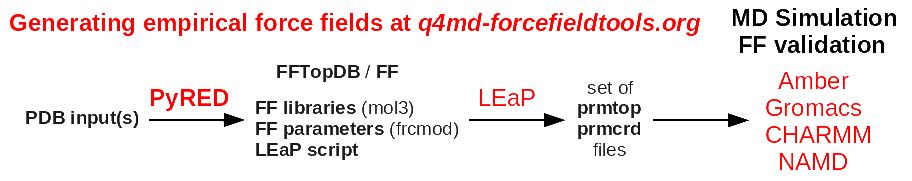
>> PyRED generate a whole force field i.e. not only atomic charges.
>>
>> See the tutorial:
>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php
>> & in particular the last ones:
>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo1.pdf
>> http://q4md-forcefieldtools.org/Tutorial/Tutorial-4-demo5.pdf
>> where you can provide your own atom types (or correct theses
>> automatically generated) and mandatory/missing force field parameters...
>>
>> the mol3 file format is the default file format for FF libraries:
>> http://q4md-forcefieldtools.org/Tutorial/leap-mol3.php
>> (it is advantageous for molecular fragments)
>>
>> regards, Francois
>>
>>
>>
>> > I see that the Carlson group has some ADP and ATP parameters that have
>> been
>> > developed for use with ff99SB. However, from my understanding, ff12SB or
>> > ff14SB are now recommended for protein simulation due to some
>> improvements
>> > in how they treat protein secondary structure. Is anybody aware of
>> whether
>> > the Carlson ADP/ATP parameters have been vetted against ff12SB or ff14SB,
>> > or if one should expect the modifications that went into those
>> force-fields
>> > to break anything badly if they are used with the Carlson ADP/ATP
>> > parameters? I'm curious because I saw a recent (2013) nice paper from the
>> > Hummer group on F1-ATPase where they still used the combination of ff99SB
>> > with these Carlson parameters.
>> >
>> > Also, does anyone know if there are any parameters for HPO42- or H2PO4-
>> > floating around out there that have been tested with Carlson ADP +
>> > ff99SB/ff12SB/ff14SB? If not then I'll go with the antechamber + RED
>> server
>> > route.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Wed Mar 04 2015 - 01:00:02 PST
{kind=link}
{kind=link}
