> I see that the Carlson group has some ADP and ATP parameters that have been
> developed for use with ff99SB. However, from my understanding, ff12SB or
> ff14SB are now recommended for protein simulation due to some improvements
> in how they treat protein secondary structure. Is anybody aware of whether
> the Carlson ADP/ATP parameters have been vetted against ff12SB or ff14SB,
> or if one should expect the modifications that went into those force-fields
> to break anything badly if they are used with the Carlson ADP/ATP
> parameters? I'm curious because I saw a recent (2013) nice paper from the
> Hummer group on F1-ATPase where they still used the combination of ff99SB
> with these Carlson parameters.
>
> Also, does anyone know if there are any parameters for HPO42- or H2PO4-
> floating around out there that have been tested with Carlson ADP +
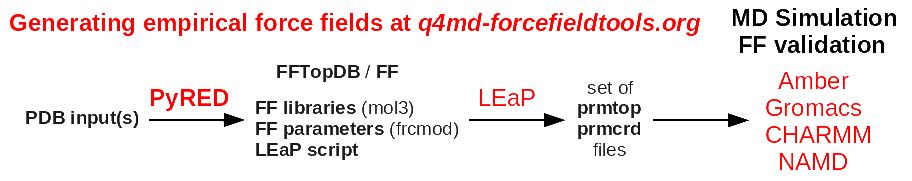
> ff99SB/ff12SB/ff14SB? If not then I'll go with the antechamber + RED server
> route.
{kind=link}
{kind=link}
