Date: Wed, 11 Feb 2015 02:07:13 -0500
Dear AMBER users,
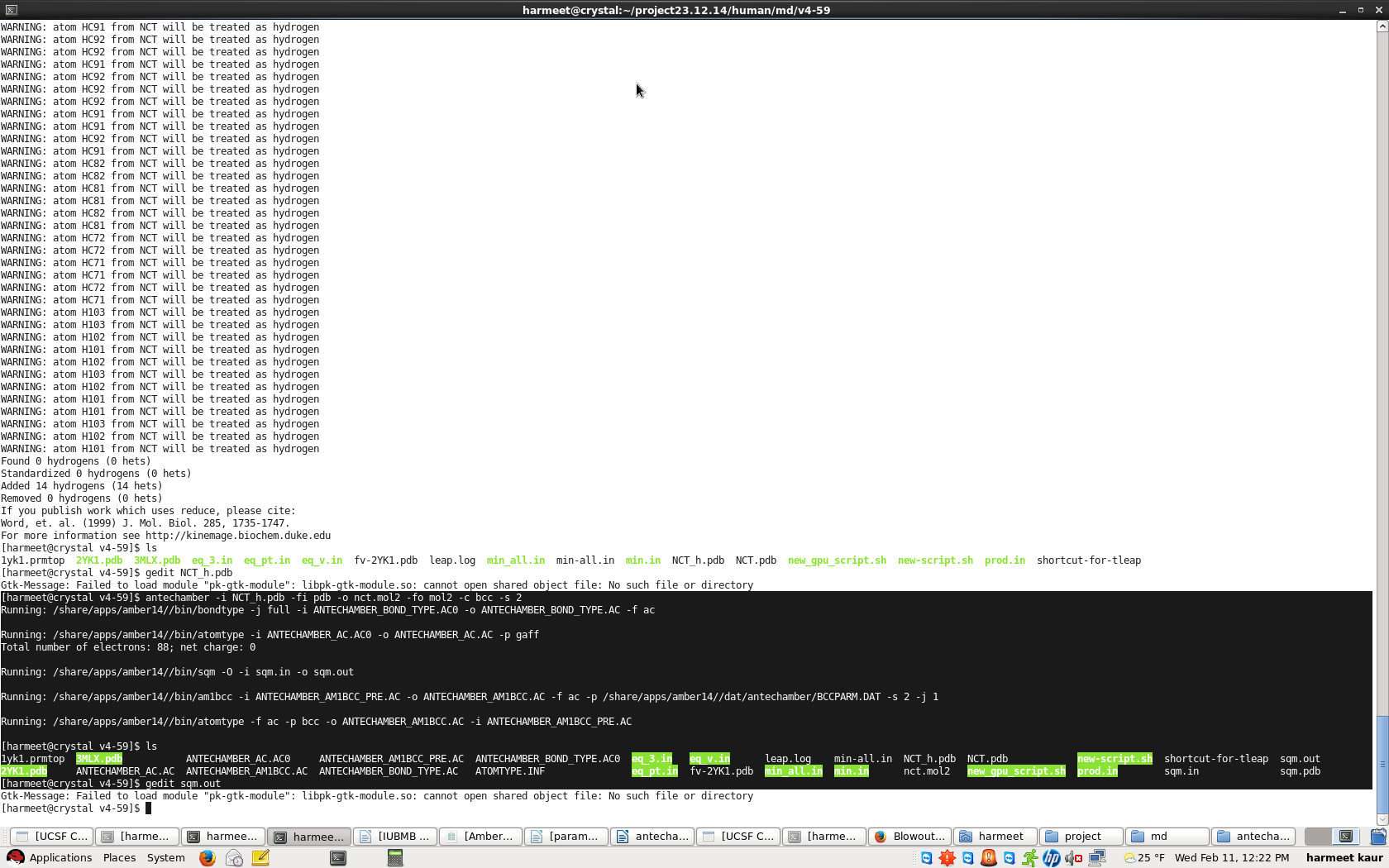
I have used antechamber to generate parameters for nicotine. However after
running the following command
*antechamber -i nct_h.pdb -fi pdb -o nct.mol2 -fo mol2 -c bcc -s 2*
I do not get mopac.xxx files, rather sqm.xxx files. It seems, that mopac.sh
isn't loaded and run, instead sqm runs. Kindly suggest why this is so. I
have attached a snapshot as reference for the same ( highlighted in black).
Thanks and regards
Harmeet
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot-3.png)
