Date: Tue, 20 Jan 2015 10:43:25 -0700
Hi,
Thanks for the PDB files. I think I may have a handle on what's going
on here. I'll start with some of your initial findings:
>> Using cpptraj (fully patched AmberTools14), I then compute the principal
>> axes of each cluster. I expected that I obtain the same results. Instead if
>> I view the resulting principal axes, overlayed with the output cluster
>> structures (as obtained from the dorotation keyword), I see that the axes
>> are indeed identical, but the clusters are offset. I have also tried the
Your original posted input was this:
trajin 4mer-1.leap.pdb
center :1-4 origin
principal :1-4 dorotation mass
trajout 4mer-1.mol2
vector v1 :1-4 principal x out 4mer-1.x.dat
...
The reason your 4mer-1.x.dat etc files containing principal axis
vector coordinates were the same for both systems for this particular
input is that the 'principal' command is actually rotating system
coordinates so that the principal axes are aligned with the coordinate
axes. So by the time you hit the 'vector' command you're actually
calculating the principal axes of the already-aligned system.
However, there is definitely something more interesting going on here.
Using the PDB files where you made the atom ordering consistent
between the two structures (thanks for that), I RMS-fit the second to
the first (after centering on the origin) and calculated the principal
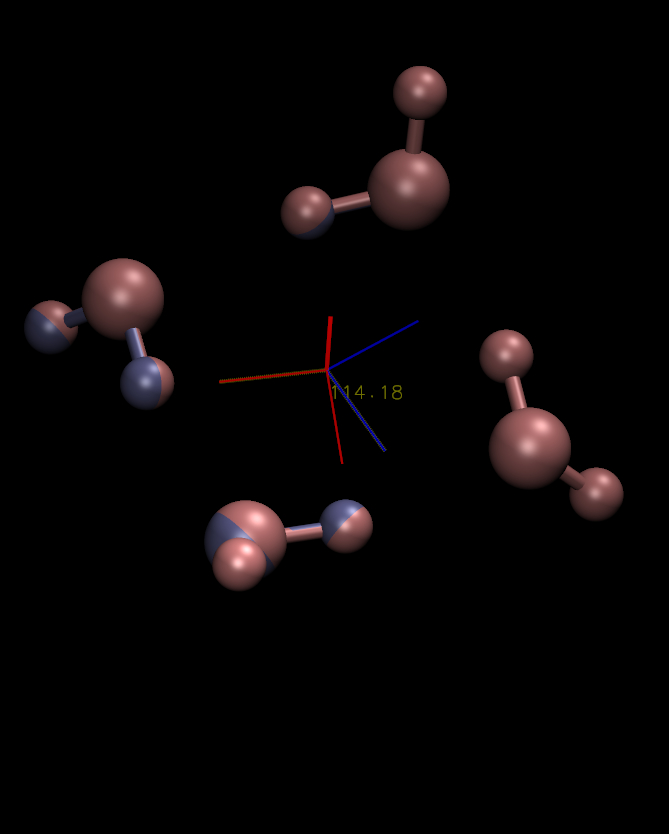
axes for both. I'm attaching a plot to try to make my subsequent
observations more clear; in it the blue-colored objects pertain to the
first PDB and the red-colored objects to the second PDB. The two
structures are indeed almost identical (RMSD of 0.0006 Ang. as you
also found) and overlap quite well. The first principal axes
(associated with largest eigenvalue, thickest lines, labelled X due to
historic convention from ptraj) from both structures overlap almost
completely as well (the angle between them is less than 0.1 degrees).
However, the second and third principal axes are offset by a rotation
of ~114 degrees around the X axis.
I'm certain that the code itself is working ok. I went through the
moment of inertia calculation step by step to ensure it was being done
properly, and used several different outside eigenvector/eigenvalue
solvers on the inertia matrix to make sure nothing weird was going on
there either (this is actually why it has taken me so long to get back
to you on this).
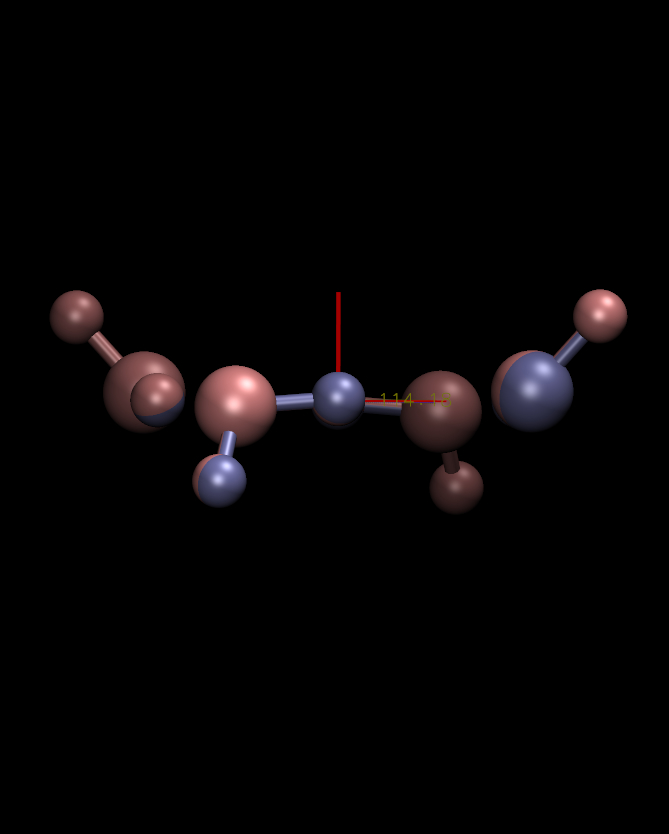
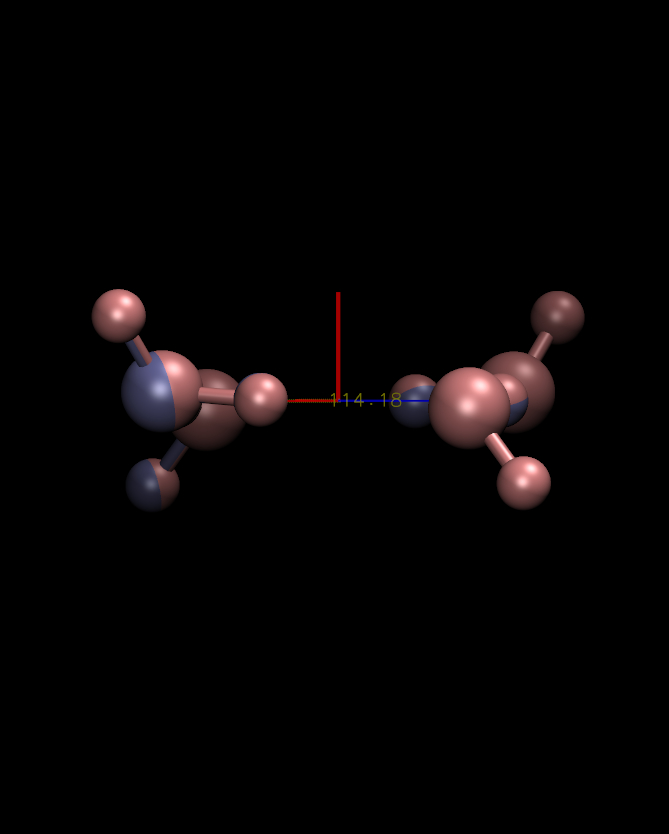
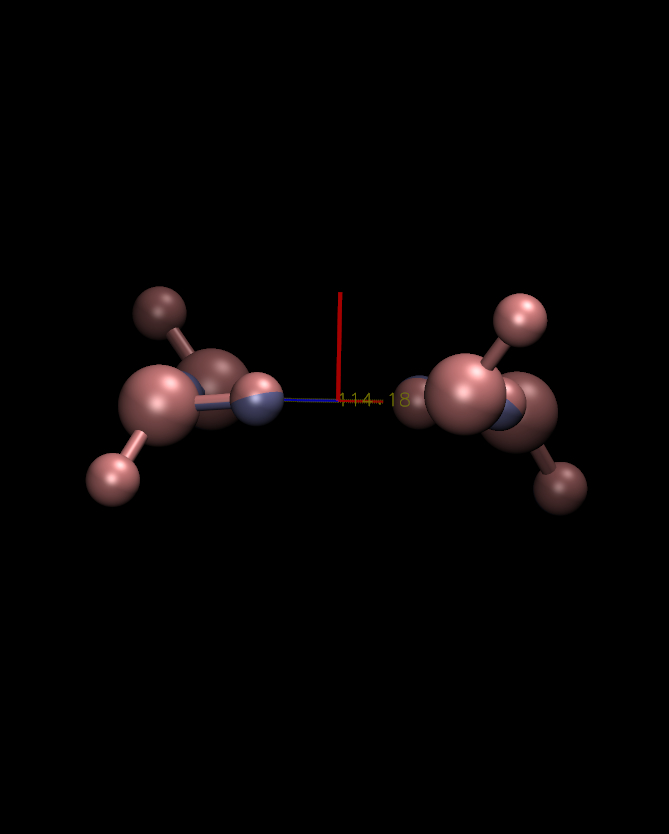
I *think* what's going on here is there are some interesting
coordinate symmetries in this system, and the slight differences in
coordinates are causing different symmetries to dominate the secondary
(Y and Z) principal axes. If you look at the eigenvalues for these
axes they are almost all degenerate (152.90 and 152.87 in the first
case vs 152.88 and 152.86 in the second). I'm attaching plots roughly
looking down the Y and Z principal axes for each structure. In each
case you appear to get a certain type of symmetry. So really none of
these answers seem wrong. To put it inelegantly, you just end up
having two degenerate sets of Y-Z axes to choose from.
Hopefully this helps make things more clear. If not (or if you don't
agree) let me know and we can chat about it further.
-Dan
>> "origin" keyword (both before and after the principal command) to help
>> correct the structures' offset, but this doesn't change the final
>> coordinates within the mol2 file. As seen in the image, the 4mer-2 has two
>> of its principal axes pointing directly at hydrogen atoms, while the 4mer-1
>> has the same two axes pointing to the center of hydrogen bonds.
>
> The 'center' command should only be necessary prior to the rotation
> from 'principal'.
>
>> I would appreciate if someone could point out if I have made an error in
>> my input or in my thinking. My only guess(!) is that perhaps something is
>> going wrong with dorotation within the principal command.
>
> Nothing appears wrong with your input at first glance. One thing to
> try is to use the 'out <filename>' keyword of the 'principal' command
> to ensure you're indeed getting the same eigenvectors (althought the
> 'principal' and 'vector' commands use the same code for calculating
> principal axes so it should match). If you would like, send me
> off-list the PDBs for each cluster and I can take a closer look.
>
> -Dan
>
>>
>> Just to be clear, the attached image shows the output structures (eg.
>> 4mer-1.mol2) and principal axes (e.g. 4mer-1.x.pdb) as obtained from
>> cpptraj, whose input for the 4mer-1 is below. The two clusters would
>> overlay perfectly if one did it manually within a molecular viewer - by an
>> ~90 degree rotation of the blue cluster.
>>
>> cpptraj.in:
>>
>> trajin 4mer-1.leap.pdb
>> center :1-4 origin
>> principal :1-4 dorotation mass
>> trajout 4mer-1.mol2
>> vector v1 :1-4 principal x out 4mer-1.x.dat
>> vector v2 :1-4 principal y out 4mer-1.y.dat
>> vector v3 :1-4 principal z out 4mer-1.z.dat
>> vector v4 :1-4 principal x trajout 4mer-1.x.pdb trajfmt pdb
>> vector v5 :1-4 principal y trajout 4mer-1.y.pdb trajfmt pdb
>> vector v6 :1-4 principal z trajout 4mer-1.z.pdb trajfmt pdb
>>
>> Thanks in advance,
>> Karl
>>
>> --
>> Karl. N. Kirschner, Ph.D.
>> Research Associate
>> Bonn-Rhein-Sieg University of Applied Sciences
>> Grantham-Allee 20, 54757 Sankt Augustin, Germany
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
>
>
> --
> -------------------------
> Daniel R. Roe, PhD
> Department of Medicinal Chemistry
> University of Utah
> 30 South 2000 East, Room 307
> Salt Lake City, UT 84112-5820
> http://home.chpc.utah.edu/~cheatham/
> (801) 587-9652
> (801) 585-6208 (Fax)
-- ------------------------- Daniel R. Roe, PhD Department of Medicinal Chemistry University of Utah 30 South 2000 East, Room 307 Salt Lake City, UT 84112-5820 http://home.chpc.utah.edu/~cheatham/ (801) 587-9652 (801) 585-6208 (Fax)
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: principalAxes.jpg)
(image/jpeg attachment: red1.jpg)

(image/jpeg attachment: red2.jpg)
(image/jpeg attachment: blue1.jpg)
(image/jpeg attachment: blue2.jpg)
