Date: Tue, 14 Oct 2014 14:44:30 -0600
Hi,
On Tue, Oct 14, 2014 at 5:25 AM, Asmita Gupta <asmita4des.gmail.com> wrote:
> This is a very old post from amber mailing list which is apparently
> unanswered..
>
> http://archive.ambermd.org/200107/0059.html
This post isn't just old, it's ancient in terms of Amber (13 years ago
we're talking Amber 6)...
> I have similar doubts as to the the calculation of principal axes in
> cpptraj. Shouldn't the vectors x,y,z be mutually perpendicular to each
> other or align with the coordinate axes of the molecule. I have used
> similar script for calculation but the vectors don't seem to be correct..
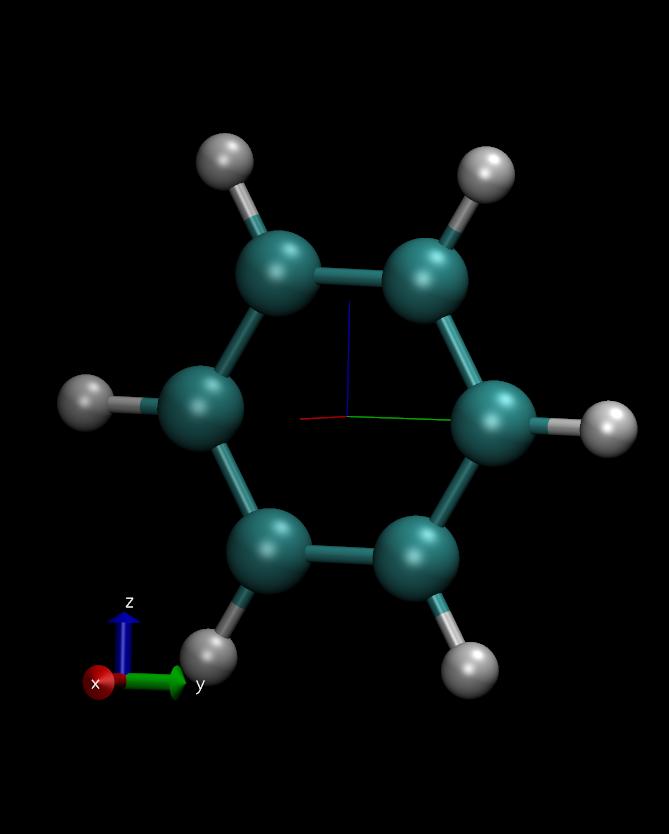
I did a quick test using benzene (since the original post mentions
using a ring structure) and the results seem fine to me (picture
attached). The picture shows the results after performing 'principal
dorotation' and 'vector principal x' (red), 'vector principal y'
(green), and 'vector principal z' (blue). The structure is aligned and
the principal axes are orthogonal to one another as one would expect.
In cpptraj (as in ptraj before it) the principal axes are (sorted from
largest to smallest eigenvalue) X > Y > Z in both the 'principal' and
'vector principal' commands. Note that this is opposite the usual
convention, which assigns the axis with the largest eigenvalue to Z;
this may have been what confused the original poster. This convention
has been maintained in cpptraj for backwards compatibility.
> Can you please suggest how can i get the 3 principal axes vectors which are
> aligned w.r.t. coordinate axes..
Something like this would work:
parm BNZ.parm7
trajin BNZ.rst7
# Align principal axes to coordinate axes
principal dorotation
# Output rotated structure
trajout rotated.mol2
# Output principal axes vectors as PDB files
vector PX principal x trajout PX.vectraj trajfmt pdb
vector PY principal y trajout PY.vectraj trajfmt pdb
vector PZ principal z trajout PZ.vectraj trajfmt pdb
Hope this helps,
-Dan
>
> Thanks
>
> Asmita
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
-- ------------------------- Daniel R. Roe, PhD Department of Medicinal Chemistry University of Utah 30 South 2000 East, Room 307 Salt Lake City, UT 84112-5820 http://home.chpc.utah.edu/~cheatham/ (801) 587-9652 (801) 585-6208 (Fax)
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: BenzenePrincipal.jpg)
