Date: Tue, 3 Jun 2014 13:20:05 -0600
Hi,
I've received your files and was able to run the remlog analysis
successfully. There does not appear to be a bug; the output from the
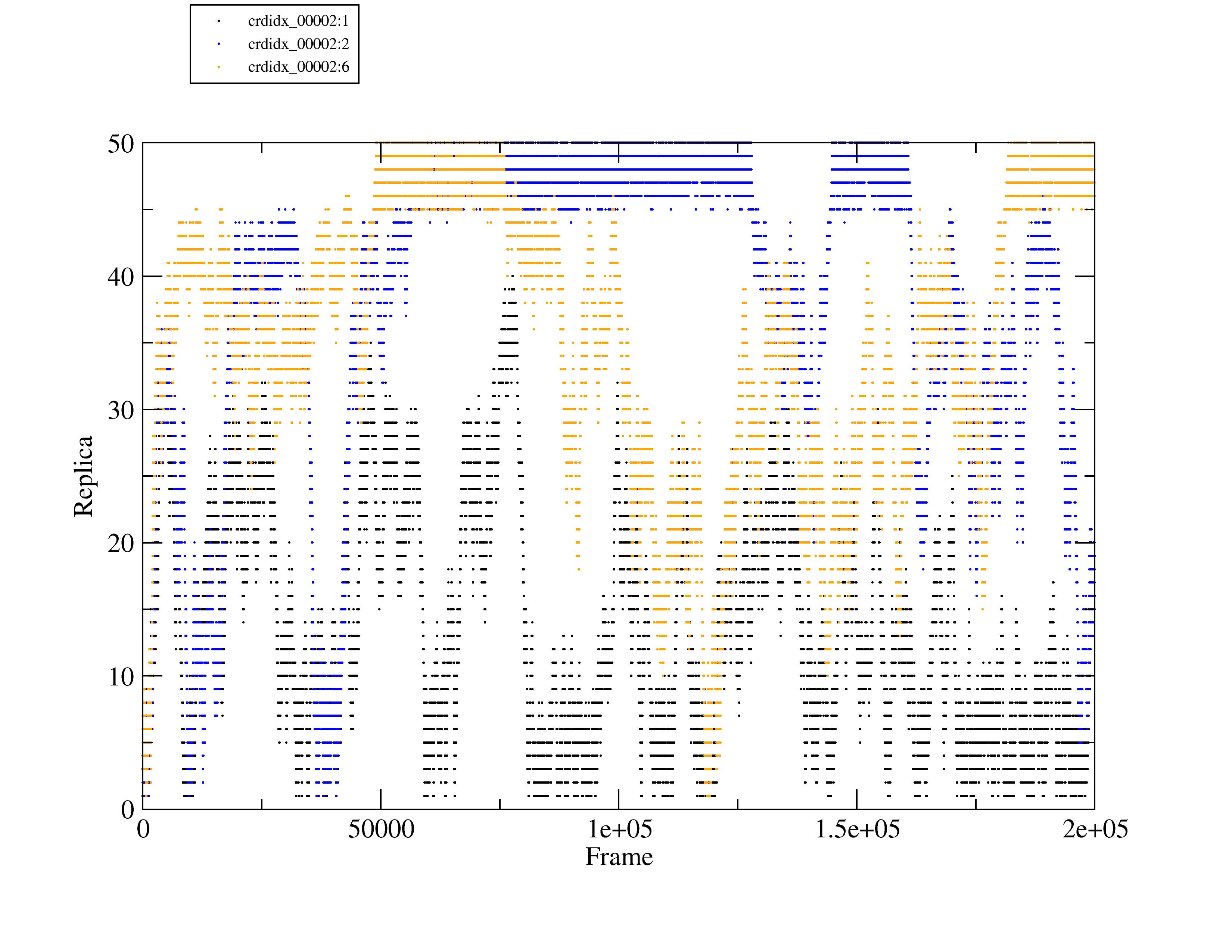
remlog analysis is accurate for your system. If you look at the
replica index vs time plot from the remlog analysis ('out <filename>
repidx') you can see that most replicas never actually complete a full
round trip (lowest->highest->lowest); I've attached a plot showing 3
of your replicas as an example. Coordinate index (CrdIdx) 1 starts out
at the bottom but never makes it to the highest replica. CrdIdx 2
starts out at the bottom and hits the highest replica, but never makes
it all the way back down again. CrdIdx 6 does actually make a round
trip (from exchange 708 to 118608); you can see individual round trips
printed by specifying 'printtrips'.
Just from the brief glance I had of your data, what appears to happen
most often is replicas tend not to reach your highest replica before
heading back down to the lowest replica (CrdIdx 5, 7, 8, 9 etc). Based
on the overall exchange acceptance calc (not yet released in cpptraj
but might be as an update later) there are a few places where your
exchange acceptance drops. Your exchange acceptance up drops from
26.97% for replica 6->7 to 10.52 for replica 7->8, and only gradually
climbs back up to 19% at replica 27. From replica 29->30->31->32->33
the acceptance ranges from 8 to 9%, and from 44->45 and 45->46 the
acceptance is 6.98% and 9.98% respectively. Any of these could act as
potential impediments for any given set of coordinates attempting to
traverse your replicas. I have found that in general 1 or two
instances of somewhat low acceptance do not seem to hurt you too
badly, but many (especially consecutive) instances of low acceptances
or an instance of extremely low acceptance (like the 6% one) will
start to have a noticeable impact. Another way to view this is to look
at the average time each set of coordinates tends to spend at each
replica ('reptime <filename>'). Ideally these lines should be flat
(i.e. coordinates are spending about the same amount of time at each
replica), but in your case certain coordinates spend way more time at
some replicas compared to others.
I'm not sure how you arrived at your temperature spacing but you may
want to try and revisit those sections in particular to try and get a
more uniform exchange acceptance %.
Hope this helps. Let me know if you have any more questions,
-Dan
On Mon, Jun 2, 2014 at 8:54 PM, Daniel Roe <daniel.r.roe.gmail.com> wrote:
> The remlog command has worked fine for me in the tests that I've
> performed for it. It won't yet work for MREMD logs, but since you're
> getting reptime etc output I don't think that's your issue. Can you
> send me off-list your file so I can try to reproduce the problem?
> Thanks.
>
> -Dan
>
> On Mon, Jun 2, 2014 at 6:25 PM, Eugene Yedvabny <eyedvabny.berkeley.edu> wrote:
>> Hello Amber community,
>>
>> I am trying to use the new cpptraj remlog command to extract the roundtrip
>> times and % exchange of my T-REMD rounds. While the 'out' and 'reptime'
>> parameters work just fine, the 'stats' parameter prints out a file full of
>> 0s for all the fields (except CRDIDX). The commands I am using is:
>>
>> readdata ../mdout/remd1.log name log1
>> remlog log1 stats statsout ../analysis/remd1_stats.dat
>>
>> Is this a known bug in cpptraj or am I somehow misusing the command?
>>
>> Thank you,
>> Eugene Yedvabny
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>
>
>
> --
> -------------------------
> Daniel R. Roe, PhD
> Department of Medicinal Chemistry
> University of Utah
> 30 South 2000 East, Room 201
> Salt Lake City, UT 84112-5820
> http://home.chpc.utah.edu/~cheatham/
> (801) 587-9652
> (801) 585-6208 (Fax)
-- ------------------------- Daniel R. Roe, PhD Department of Medicinal Chemistry University of Utah 30 South 2000 East, Room 201 Salt Lake City, UT 84112-5820 http://home.chpc.utah.edu/~cheatham/ (801) 587-9652 (801) 585-6208 (Fax)
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: repidx.jpg)
