Date: Thu, 08 May 2014 08:22:41 -0400
On Thu, 2014-05-08 at 11:16 +0100, Hannes Loeffler wrote:
> Hi,
>
> I can't get the attached structure (sqm.in) converged. What happens is
> that the wave function won't converge somewhere along geometry
> optimisation, at some arbitrary step (not the first step) depending on
> name list parameters.
>
> I have no particular interest in this one. The problem just came up in
> a larger test set. The structure is ZINC03814828. Same result with
> ZINC03814832 which is almost the same.
>
> The current name list is an outcome of playing around with various
> parameters but without luck. What I wonder is if this could still be
> made to converge or if there is a set of known structures which just
> cannot be converged? If I replace the carboxy-ethyl group with propyl
> the structure converges at scfconv=1.0D-9.
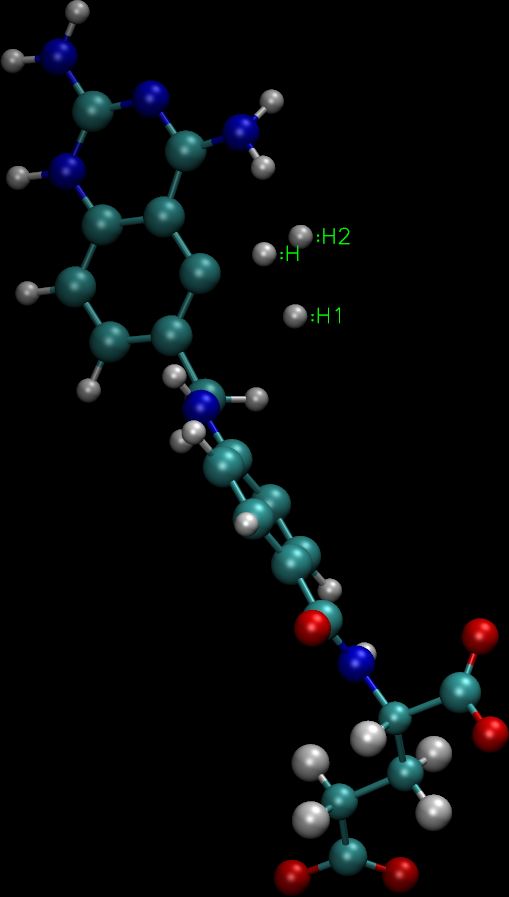
I think I may be missing something obvious or perhaps I postprocessed
your sqm input file wrong. If I delete the sqm input variables and put
'55' at the top of the file (to try and make the file look like an XYZ),
I get an image which appears to have 3 hydrogen atoms floating around
somewhere in space (image attached).
If I didn't make a mistake in my hacky sqm-to-xyz file conversion (it is
_very_ possible that I did), this could certainly explain why the
structure fails to converge (most of the structure looks fine, but not
the 3 labeled H atoms), and more reflects a problem with the starting
structure than with sqm.
All the best,
Jason
-- Jason M. Swails BioMaPS, Rutgers University Postdoctoral Researcher
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: zinc03814828.png)
