Marianna,
and Yes you can use the drawing mode of XLEaP to create your molecule...
just create the heavy atom in dirty & quich way (linear) and then "add
hydrogen and built " should lead to a starting structure; save the
file using the 'savepdb' command
regards, F.
----- Forwarded message from fyd.q4md-forcefieldtools.org -----
Date: Wed, 07 May 2014 17:13:05 +0200
From: FyD <fyd.q4md-forcefieldtools.org>
Reply-To: FyD <fyd.q4md-forcefieldtools.org>
Subject: Re: [AMBER] PDB for 1-octanol molecules
To: AMBER Mailing List <amber.ambermd.org>
Dear Marianna,
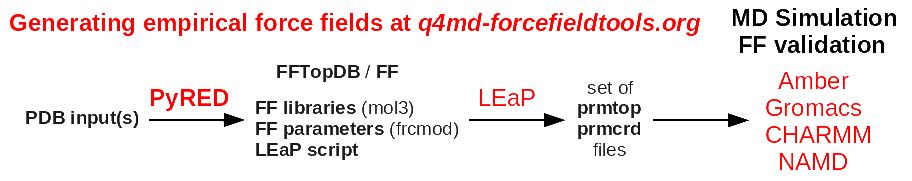
You first need to generate a force field for 1-octanol.
You can use
http://q4md-forcefieldtools.org/REDServer-Development/ to
generate a force for that molecule; at the end you end up with a
leaprc.q4mdfft file that you load in the LEaP problem (AmberTools)
See
http://q4md-forcefieldtools.org/REDServer-Development/images/PyRED.gif
http://q4md-forcefieldtools.org/Tutorial/Tutorial-4.php#6
then you will be able to solvate 1-octanol by itself in LEaP...
regards, Francois
> I would like to build a box of 1-octanol molecules and use the GAFF force
> field to perform MD simulations. I have gone through several of the
> tutorials available online (enormously helpful, by the way) and had success
> running MD on a single molecule of 1-octanol.
>
> I have built pdb files for boxes of octanol molecules using other programs,
> but I was unable to get Amber to read these files properly. Can Xleap, or
> any of the other utilities within Amber, be used to build a pdb file
> corresponding to a box of several (e.g., a few hundred) octanol molecules?
----- End forwarded message -----
F.-Y. Dupradeau
---
http://q4md-forcefieldtools.org/FyD/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Wed May 07 2014 - 08:30:07 PDT
{kind=link}
