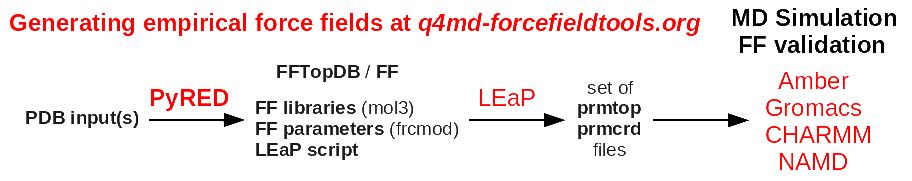
> I want to simulate Glycine in vacuum with amber,what I done like this:
>
> tleap ?s ?f $AMBERHOME/dat/leap/cmd/leaprc.ff99SB
> a = sequence{GLY}
> saveamberparm a gly.prmtop gly.inpcrd
>
> but I found that Gly residue in amber is NH-CH2-CO which reduces a -H and a
> -OH compared with the real Glycine like NH2-CH2-COOH in the shape of solid
> powder , so how can I add these two parts to generate a Glycine complete
> prmtop file?
{kind=link}
