Date: Mon, 24 Mar 2014 08:31:25 +0500
*Dear Amber Experts, *
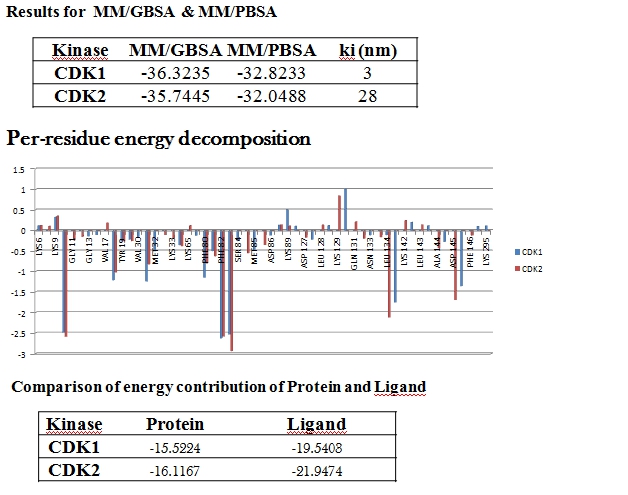
I am running MD simulation for a protein ligand complex to determine
selectivity. When I calculate the binding free energy, the MMGBS value is
according to the published data. But when I perform energy decomposition
using MMGBSA.py (Amber Tools 13) to calculate per residue contribution, all
of the energy shift difference shifts toward ligand, and the contribution
of amino acids become opposite.
Data for the comparison of MMGBSA and decomposition is attached along with
mail. Please guide me to solve this problem.
-- *Regards,* *Tahir Ali Chohan* *B.Pharm., M.Phil (Pharm. Chem.)* *PhD Scholar* *College of Pharmaceutical Sciences* *Zhejiang University, Hangzhou, China.* *Cell # 0086-13018996850*
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: image-03d29dee3a12d0e63885b3e08529208f8c4b6462d875c2dd7f3ccf3d015bd5fb-V_1_.jpg)
