Date: Tue, 5 Nov 2013 11:31:54 +0100 (CET)
Hello,
I did a quick test on the force field that Hector provided using Wolf2Pack. The attached n3-s6-ca_benezesulfanamide.png indicates that the angle parameter does not reproduce the HF/6-31G(d) minima (i.e. 104.7 degrees, blue curve). By adjusting the angle value to 102.0 in the force field file, you obtain good agreement as shown in n3-s6-ca_opt_benezesulfanamide.png.
The generic torsion parameter x-n3-s6-x does not model the hn-n2-s6-ca rotation very well. The global minima of this curve is reproduced at 240 degrees, but the second low energy minima (HF/6-31G(d) = 1.03 kcal/mol) at 60 degrees is a transition state according to the molecular mechanics curve, as seen in x-n3-s6-x_benezesulfanamide.png (1 = 0 degrees, 12 = 30 degrees). To correct this curve, one would need to decouple the hn-n2-s6-ca from the hn-n2-s6-o term.
Because of the torsion curve shown, I would suggest not using the generic x-n3-s6-x force field term - especially if the rotation about the central bond is of importance for your investigation. (There may be additional parameters that could be optimized, but I didn't check their performance. Examples of this may include n3-s6-o or n3-s6-ca.) You are welcomed to check these yourself using W2P.
Best Regards,
Karl
------------------------------------
Karl N. Kirschner, Ph.D.
Fraunhofer-Institute for Algorithms
and Scientific Computing - SCAI
Department of Bioinformatics
53754 Sankt Augustin, Germany
Tel: +49 (0) 2241-14-2052
Fax: +49 (0) 2241-14-1328
------------------------------------
----- Original Message -----
From: "Hector A. Baldoni" <hbaldoni.unsl.edu.ar>
To: "AMBER Mailing List" <amber.ambermd.org>
Sent: Monday, November 4, 2013 11:00:49 PM
Subject: Re: [AMBER] Sulfonamide parameters,
Hi!
The attached frcmod file will give you a nice shaped sp3 to you
sulfonamide moiety. I checked it in 2FOU pdb file.
Greeting,
Hector.
> *Dear experts,*
>
> I am interested in molecular dynamics simulation with small molecule and
> protein to explore selectivity mechanism. But unfortunately I have
> encountered some problem and I need help.
>
> 1). The ligand I am using, contains Benzene sulfonamide group.
>
> 2) As I perform the energy minimization the angles of the H-N-S and N-S-O
> bonds becomes unnatural, both of which are a little over 90 degrees.
>
> 3) I am using the following method for calculation of charges of my
> ligands
> (p hf/6-31g(d) pop=mk iop(6/33=2).
>
> During my search on Google I found that some other people also have been
> suffered with the same problem and finally they solved it by making some
> modification in their input files.
>
>
>
> So I need help to fix out this problem.
>
> I will be grateful for this favor.
> Here I am attaching original PDB of my ligand, Mol2 file, PDB after energy
> minimization (min3) and frcmod file. So that you can give me batter
> suggestion.
>
> --
> *Regards,*
> *Tahir Ali Chohan*
> *B.Pharm., M.Phil (Pharm. Chem.)*
> *PhD Scholar*
> *College of Pharmaceutical Sciences*
> *Zhejiang University, Hangzhou, China.*
> *Cell # 0086-13018996850*
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
--------------------------------------
Dr. Hector A. Baldoni
Area de Quimica General e Inorganica
Universidad Nacional de San Luis
Chacabuco 917 (D5700BWS)
San Luis - Argentina
hbaldoni at unsl dot edu dot ar
Tel.:+54-(0)266-4520300 ext. 6157
--------------------------------------
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
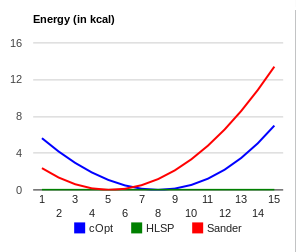
(image/png attachment: n3-s6-ca_benezesulfanamide.png)
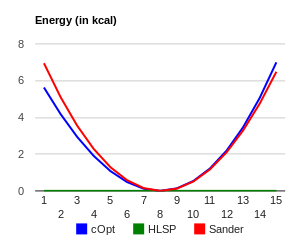
(image/png attachment: n3-s6-ca_opt_benezesulfanamide.png)
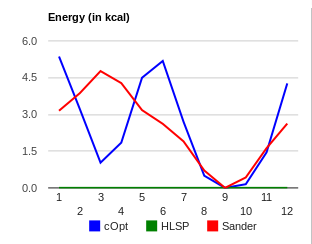
(image/png attachment: x-n3-s6-x_benezesulfanamide.png)
