Date: Thu, 19 Sep 2013 15:06:44 -0400
On Thu, Sep 19, 2013 at 2:49 PM, Ray Luo, Ph.D. <ray.luo.uci.edu> wrote:
> The complex internal energy terms are:
>
> GENERALIZED BORN:
>
> WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIAL
> TERMS. THE VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> Complex:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
>
> -------------------------------------------------------------------------------
> BOND 1046.0599 25.0584
> 3.5438
> ANGLE 2825.6096 27.8852
> 3.9436
> DIHED 3867.0393 20.6518
> 2.9206
> VDWAALS -2766.8957 18.9666
> 2.6823
> EEL -22450.1018 52.8100
> 7.4685
> 1-4 VDW 1296.0772 16.0482
> 2.2696
> 1-4 EEL 14608.9084 34.7009
> 4.9074
> EGB -4636.7135 33.1652
> 4.6903
> ESURF 146.7058 1.1452
> 0.1620
>
> G gas -1573.3031 55.6723
> 7.8733
> G solv -4490.0077 33.3028
> 4.7097
>
> TOTAL -6063.3108 43.9275
> 6.2123
>
> Similar for the Poisson-Boltzmann part. There is an image in that
> email too. Not sure you got that picture ...
>
I did get the image, but that is not a large internal/gas-phase energy for
that system. This is a 358 residue protein. With one of my systems, 129
residues, this is one of my MD records (implicit solvent):
NSTEP = 6000 TIME(PS) = 603011.997 TEMP(K) = 307.92 PRESS =
0.0
Etot = -2437.0929 EKtot = 1528.2048 EPtot =
-3965.2977
BOND = 404.1507 ANGLE = 1005.0443 DIHED =
1344.4584
1-4 NB = 412.2395 1-4 EEL = 3681.8749 VDWAALS =
-946.8635
EELEC = -6006.1019 EGB = -3860.1002 RESTRAINT =
0.0000
------------------------------------------------------------------------------
Extending this to a protein ca. 3x bigger, I think the internal (and
non-bonded) energy terms fall in the right ballpark. My suspicion is that
there was some mistake in the visualization of the ligand structure (I did
see the image). An image that warped would simply not give energies that
look even close to reasonable.
Moreover, since the internal potential terms cancel for all terms except
the dihedral (which leads me to wonder why not -- that is another problem),
means that any issues in the complex must be present also in the receptor
or ligand (and it's the ligand energy terms in the unbound state that is
giving rise to NaNs).
At the moment I can't say what went wrong...
All the best,
Jason
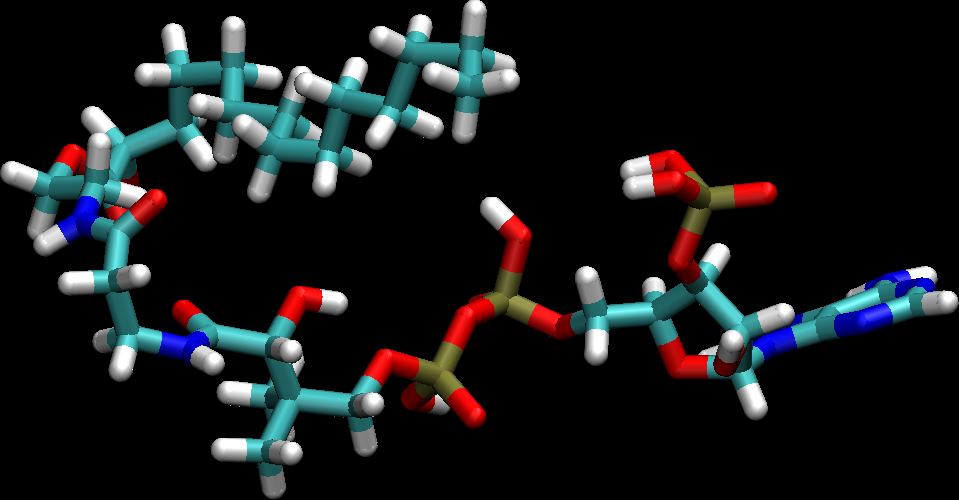
P.S. The image I got when I visualized the OP's ligand prmtop with the
ligand trajectory file is attached -- looks quite reasonable.
-- Jason M. Swails BioMaPS, Rutgers University Postdoctoral Researcher
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: molecule.png)
