Date: Thu, 19 Sep 2013 18:05:08 -0700
Hello,
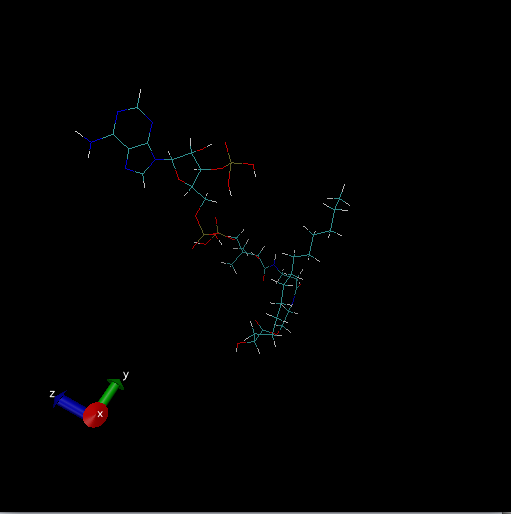
Looking at the image of your ligand, there is definitely something odd.
Are you using periodic boundaries when you load the ligand into VMD for
viewing? When I loaded the ligand from the files you sent into VMD using
Coordinates With Periodic Box info, it gives an image like you show.
When I load without Periodic Box info, it shows up just fine.
I will see if I can replicate the NaN error under PBSA itself to see if
the source of the problem is there or something else.
-Wes
> Dear Jason
> My input file consists of following parameters:
> Input file for running PB and GB
> &general
>
> startframe=1, endframe=50, interval=1,
>
> verbose=2, keep_files=0,
>
> /
> &gb
>
> igb=5, saltcon=0.150,
>
> /
> &pb
>
> istrng=0.15, fillratio=4.0
> /
>
> the command i used is:
> MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp
> amcligsolvated.prmtop -cp complex.prmtop -rp protein.prmtop -lp
> ligand.prmtop -y amcprod1.mdcrd
> command ran smooth no error appeared on terminal but when i checked output
> file i came to know that NAN error in case of ligand occured because of
> which no energy difference calculated.
> Output was:
>
> | Run on Tue Sep 17 17:20:17 2013
> |
> |Input file:
> |--------------------------------------------------------------
> |Input file for running PB and GB in serial
> |&general
> | endframe=50, keep_files=2,
> |/
> |&gb
> | igb=2, saltcon=0.100,
> |/
> |&pb
> | istrng=0.100,
> |/
> | --------------------------------------------------------------
> |MMPBSA.py Version=12.0
> |Solvated complex topology file: amcligsolvated.prmtop
> |Complex topology file: amcliguUS.prmtop
> |Receptor topology file: pro.prmtop
> |Ligand topology file: unk.prmtop
> |Initial mdcrd(s): amcprod1.mdcrd
> |
> |Receptor mask: ":1-357"
> |Ligand mask: ":358"
> |Ligand residue name is "UNK"
> |
> |Calculations performed using 50 complex frames.
> |Poisson Boltzmann calculations performed using internal PBSA solver in
> mmpbsa_py_energy
> |
> |All units are reported in kcal/mole.
> -------------------------------------------------------------------------------
> -------------------------------------------------------------------------------
>
> GENERALIZED BORN:
>
> WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIAL
> TERMS. THE VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> Complex:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 1046.0599 25.0584
> 3.5438
> ANGLE 2825.6096 27.8852
> 3.9436
> DIHED 3867.0393 20.6518
> 2.9206
> VDWAALS -2766.8957 18.9666
> 2.6823
> EEL -22450.1018 52.8100
> 7.4685
> 1-4 VDW 1296.0772 16.0482
> 2.2696
> 1-4 EEL 14608.9084 34.7009
> 4.9074
> EGB -4636.7135 33.1652
> 4.6903
> ESURF 146.7058 1.1452
> 0.1620
>
> G gas -1573.3031 55.6723
> 7.8733
> G solv -4490.0077 33.3028
> 4.7097
>
> TOTAL -6063.3108 43.9275
> 6.2123
>
>
> Receptor:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 1023.2391 25.1818
> 3.5612
> ANGLE 2758.3780 27.3729
> 3.8711
> DIHED 3808.4462 19.5218
> 2.7608
> VDWAALS -2656.1969 18.4136
> 2.6041
> EEL -22450.1018 52.8100
> 7.4685
> 1-4 VDW 1272.7137 15.8878
> 2.2469
> 1-4 EEL 14608.9084 34.7009
> 4.9074
> EGB -4662.4389 33.1432
> 4.6872
> ESURF 148.2532 1.1910
> 0.1684
>
> G gas -1634.6133 55.0064
> 7.7791
> G solv -4514.1857 33.2875
> 4.7076
>
> TOTAL -6148.7990 44.1153
> 6.2389
>
>
> Ligand:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 22.8209 3.7323
> 0.5278
> ANGLE 67.2316 5.6725
> 0.8022
> DIHED 57.9771 3.3766
> 0.4775
> VDWAALS -21.1078 1.7341
> 0.2452
> 1-4 VDW 23.3635 2.0523
> 0.2902
> ESURF 8.4113 0.1439
> 0.0204
>
> G gas 150.2853 7.8027
> 1.1035
> G solv 8.4113 0.1439
> 0.0204
>
> TOTAL 158.6965 7.8116
> 1.1047
>
>
> Differences (Complex - Receptor - Ligand):
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND -0.0000 0.0000
> 0.0000
> ANGLE -0.0000 0.0001
> 0.0000
> DIHED 0.6160 0.3232
> 0.0457
> VDWAALS -89.5910 3.1602
> 0.4469
> EEL 0.0000 0.0000
> 0.0000
> 1-4 VDW -0.0000 0.0001
> 0.0000
> 1-4 EEL 0.0000 0.0000
> 0.0000
> EGB 25.7254 1.2092
> 0.1710
> ESURF -9.9586 0.2770
> 0.0392
>
> DELTA G gas -88.9750 3.0895
> 0.4369
> DELTA G solv 15.7667 1.1381
> 0.1610
>
> DELTA TOTAL -73.2083 3.0542
> 0.4319
>
>
> -------------------------------------------------------------------------------
> -------------------------------------------------------------------------------
>
> POISSON BOLTZMANN:
>
> WARNING: INCONSISTENCIES EXIST WITHIN INTERNAL POTENTIAL
> TERMS. THE VALIDITY OF THESE RESULTS ARE HIGHLY QUESTIONABLE
> Complex:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 1046.0599 25.0584
> 3.5438
> ANGLE 2825.6096 27.8852
> 3.9436
> DIHED 3867.0393 20.6518
> 2.9206
> VDWAALS -2766.8957 18.9666
> 2.6823
> EEL -22450.1018 52.8100
> 7.4685
> 1-4 VDW 1296.0772 16.0482
> 2.2696
> 1-4 EEL 14608.9084 34.7009
> 4.9074
> EPB -4507.0803 33.7185
> 4.7685
> ENPOLAR 2907.4367 7.0087
> 0.9912
>
> G gas -1573.3031 55.6723
> 7.8733
> G solv -1599.6436 35.2985
> 4.9920
>
> TOTAL -3172.9466 46.1397
> 6.5251
>
>
> Receptor:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 1023.2391 25.1818
> 3.5612
> ANGLE 2758.3780 27.3729
> 3.8711
> DIHED 3808.4462 19.5218
> 2.7608
> VDWAALS -2656.1969 18.4136
> 2.6041
> EEL -22450.1018 52.8100
> 7.4685
> 1-4 VDW 1272.7137 15.8878
> 2.2469
> 1-4 EEL 14608.9084 34.7009
> 4.9074
> EPB -4549.0703 33.2881
> 4.7076
> ENPOLAR 2865.0984 7.0316
> 0.9944
>
> G gas -1634.6133 55.0064
> 7.7791
> G solv -1683.9719 34.8674
> 4.9310
>
> TOTAL -3318.5852 46.6278
> 6.5942
>
>
> Ligand:
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND 22.8209 3.7323
> 0.5278
> ANGLE 67.2316 5.6725
> 0.8022
> DIHED 57.9771 3.3766
> 0.4775
> VDWAALS -21.1078 1.7341
> 0.2452
> 1-4 VDW 23.3635 2.0523
> 0.2902
> EPB nan nan
> nan
> ENPOLAR 97.8510 0.8688
> 0.1229
>
> G gas 150.2853 7.8027
> 1.1035
> G solv nan nan
> nan
>
> TOTAL nan nan
> nan
>
>
> Differences (Complex - Receptor - Ligand):
> Energy Component Average Std. Dev. Std. Err. of
> Mean
> -------------------------------------------------------------------------------
> BOND -0.0000 0.0000
> 0.0000
> ANGLE -0.0000 0.0001
> 0.0000
> DIHED 0.6160 0.3232
> 0.0457
> VDWAALS -89.5910 3.1602
> 0.4469
> EEL 0.0000 0.0000
> 0.0000
> 1-4 VDW -0.0000 0.0001
> 0.0000
> 1-4 EEL 0.0000 0.0000
> 0.0000
> EPB nan nan
> nan
> ENPOLAR -55.5128 1.3871
> 0.1962
> EDISPER 0.0000 0.0000
> 0.0000
>
> DELTA G gas -88.9750 3.0895
> 0.4369
> DELTA G solv nan nan
> nan
>
> DELTA TOTAL nan nan
> nan
>
>
> -------------------------------------------------------------------------------
> -------------------------------------------------------------------------------
> when i visualized prmtop and MMBPSA.py generated mdcrds i found
> a distorted ligand. i also tried MMPBSA.pl but no results.
>
> kindly attached image of ligand hopefully these would be helpful for you
>
>
> Regards
>
>
>
>
>
> Sumra Wajid Abbasi
> PhD Scholar
> Computational Biology Lab, National Center for Bioinformatics
> Quaid-e-Azam University, Islamabad-45320 Pakistan.
>
>
> On Thu, Sep 19, 2013 at 3:41 PM, Jason Swails
> <jason.swails.gmail.com>wrote:
>
>> On Thu, Sep 19, 2013 at 4:15 AM, Sumra Wajid Abbasi 30-FBAS/MSBI/F09 <
>> sumra.msbi30.iiu.edu.pk> wrote:
>>
>> > Dear Jason
>> >
>> > NAN= No Atom Name
>> >
>>
>> Actually NaN means "Not a Number." [http://en.wikipedia.org/wiki/NaN]
>> It is an almost universal sign of some type of problem (either user or
>> programmer).
>>
>> EPB/EGB = the electrostatic contribution to the solvation free energy
>> > calculated by PB or GB respectively
>> > ECAVITY = nonpolar contribution to the solvation free energy
>> calculated
>> by
>> > an empirical model
>> >
>>
>> You misunderstood me. I'm aware of what all of these terms mean. What
>> is
>> important when debugging is _which_ terms had NaN as their respective
>> values? Since the solvation free energy is NaN, that means either the
>> polar solvation term (EPB/EGB) or the non-polar solvation term (ECAVITY,
>> ESURF, EDISPER) must be NaN (or both). Knowing this gives clues about
>> where to look for problems. Of course you also omitted the G gas term
>> from
>> your email, so there could be problems there as well.
>>
>> I have visualized MMPBSA.py generated mdcrds using VMD...what i feel is
>> > there are abnormalities in ligand geometry and also few in receptor..
>> >
>>
>> This does not help, as it leaves me to "guess" what the 'abnormalities'
>> are
>> (and what you would consider 'normal'). For instance:
>> http://www.mybiosoftware.com/wp-content/uploads/2011/03/AmberTools.jpg
>> and http://archive.ambermd.org/201103/att-0730/Picture_2.png are two
>> different 'abnormalities' which have quite different root causes and
>> solutions. A picture is worth more than any number of words in this
>> case.
>>
>> As general advice when asking questions of this nature, you will receive
>> helpful answers far faster if you provide a good amount of details.
>> Show
>> the full output (not just lines you find troubling, because those are
>> often
>> not helpful by themselves). Show the exact commands that you used and
>> any
>> error messages or outputs that were produced. Also say how you tried to
>> fix or debug it, and what you learned (or think you learned) in the
>> process. Otherwise, the first few days will be spent trading emails of
>> this sort...
>>
>> HTH,
>> Jason
>>
>> --
>> Jason M. Swails
>> BioMaPS,
>> Rutgers University
>> Postdoctoral Researcher
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screen_shot_2013-09-19_at_6.01.34_PM.png)

{kind=link}
{kind=link}