Date: Thu, 5 Sep 2013 14:13:16 -0500
Hi Jason and other gurus,
Really thanks for your help. We have modified the script and generate
correlograms.
So after reading several text
books<http://www.ltrr.arizona.edu/~dmeko/notes_3.pdf>,
web info<http://homepage.univie.ac.at/franz.vesely/simsp/dx/node23.html#F6ACF>
and
Amber related papers about (auto)correlation, we still have a question:
how to interpret the correlogram and know the correlation time from the
plot:
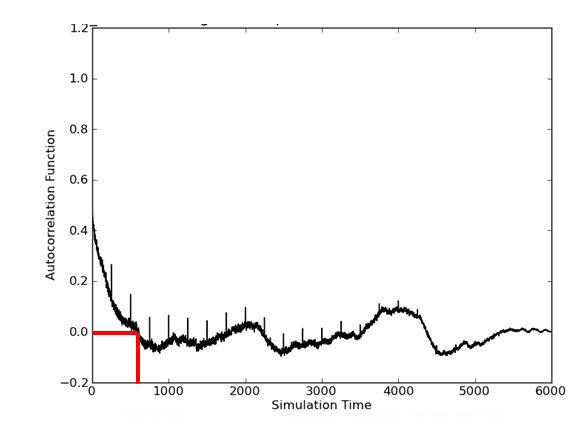
The attached is an example of the mmGBSA dG autocorrelation plot
generated from the python API. (We take one snapshot from each ps MD
trajectory. So here are one 6000 ps trajectory and 6000 snapshots of
mmGBSA calculation.) So our understanding is that during around 700 ps, as
noted in the red lines, the autocorrelation diminishes to the zero level
(the following lag from 700 ps to 6000ps fluctuates around 0 level ). So
the correlation time of this mmGBSA calculation time would be around 700
ps?
Cheers,
Henry
On Fri, Aug 30, 2013 at 12:29 PM, Jason Swails <jason.swails.gmail.com>wrote:
> On Fri, Aug 30, 2013 at 11:30 AM, <psu4.uic.edu> wrote:
>
> > Hello all,
> >
> > Thanks Jan-Philip's solution. It works! So it seems like our
> > trouble-shooting was in the wrong direction : /
> >
> > However, there is a new error message pops out:
> >
> > Loading and checking parameter files for compatibility...
> > Loading and checking parameter files for compatibility...
> > Traceback (most recent call last):
> > File "./python_API_2.py", line 21, in <module>
> > total_mut = data.mutant['gb']['complex']['TOTAL'].copy()
> > *AttributeError: 'mmpbsa_data' object has no attribute 'mutant'*
> >
>
> This example was meant primarily to demonstrate a potential use of the API.
> Attributes and data keys are created only as they're needed. Therefore,
> mmpbsa_data will not have a "mutant" attribute unless you actually run
> alanine scanning (thereby generating 'mutant' data).
>
> As general advice with examples of this sort -- analyze the code in the
> example so that you understand what each line is doing (this sample script
> just computes and plots the autocorrelation function of the total complex
> energies -- not likely of much use for what you want to do). It would be
> good to cross-reference properties of the 'data' object with how the
> AmberTools 13 manual describes the mmpbsa_data data structure. It also
> never hurts to be comfortable with using the programming language used in
> the example. FWIW, I'm hardly alone in thinking that Python programming is
> potentially the most useful
>
> A 'quick fix' for this error of course is to delete all references to the
> mutant data. However, while that will give you the satisfaction of
> watching the script finish and a graph appear, it is unlikely to provide
> you with useful data.
>
> HTH,
> Jason
>
> --
> Jason M. Swails
> BioMaPS,
> Rutgers University
> Postdoctoral Researcher
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Pin-Chih Su (Henry Su) Ph.D. canditate Center for Pharmaceutical Biotechnology (MC 870) College of Pharmacy, University of Illinois at Chicago 900 South Ashland Avenue, Room 1052 Chicago, IL 60607-7173 office 312-996-5388 fax 312-413-9303
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: correlation_time_plot.JPG)
