Date: Thu, 4 Jul 2013 09:47:32 +0900
Hello, amber users,
Now, I am reading a paper that is "Insights into structural properties of
denatured human prion 121-230 at melting temperature studied by replica
exchange molecular dynamics". Base on the describe of paper, I write the
process and the input file of amber simulation, but I cannot sure that is
correct.****
Who can help me check it?****
**
Thank a lot.
Biao Ma
**
pumed url of the paper: http://www.ncbi.nlm.nih.gov/pubmed/22339436****
** **
Blow is a part of method in the paper:****
=====start
In this work, the temperature setting used for sheep PrP 125-230 such that
MD simulations were exchanged at 320.0, 322.0,324.0, 326.0, 328.1, 330.2,
332.3, 334.4, 336.5, 338.6, 340.8, 343.0, 345.2, 347.4, 349.6, 351.8,
354.0, 356.2, 358.5, 360.8, 363.1, 365.4, 367.7, and 370.0 K was used. All
of the replicas were equilibrated for 20 ns without exchanging temperatures
and then extended for 65 ns of REMD simulation. The generalized Born model
used in this study modified the calculation of Born radii and improved the
accuracy in the solvent polarization for macromolecules. The combinational
use of the all-atom point-charge force-field (also known as ff03) and the
generalized Born model led to successful folding of several proteins. The
AMBER 11 simulation package 26 was used in both REMD simulation and data
analysis. The melted huPrP 121-230 was computed starting from an extended
huPrP. To generate the initial extended structure, a heating method was
applied to a known NMR structure (PDBcode 1hjn,15Figure 1A), enabling it to
unfold at 600 K for 40 ns of MD simulation to result in an extended
conformation (Figure 1B) as described previously.****
During this simulation, the disulfide covalent bond between residues 179
and 214 was preserved. In total, 24 replicas with duration of 65 ns and
with an integration time step of 2 fs were computed based on the extended
huPrP with different random number seeds to generate the initial
conditions. A 16 Å force-shifted non-bonded cutoff and generalized Born
solvent models with salt concentration of 0.2 M were applied.****
**=====end
**
**
**
Simulation Procedure:****
1. system building****
2. system minimization****
3. heating system****
4. generate the extend conformation****
5. local minimization after heating system****
6. equilibrate the every replica****
7. REMD simulation****
** **
** **
*1.leap.inp for system building*
** **
source pdb: 1ag2****
use the the ff03 (Duan et al.) force field****
** **
leap.inp
source leaprc.ff03.r1****
set default PBradii mbondi2****
# load pdb file****
1ag2 = loadPdb input.pdb****
savePdb 1ag2 1ag2.pdb****
bond 1ag2.179.SG <http://1ag2.179.sg/> 1ag2.214.SG <http://1ag2.214.sg/>****
# save 1ag2 to prmtop and inpcrd files****
saveAmberParm 1ag2 1ag2.prmtop 1ag2.inpcrd****
# finish****
quit****
** **
2. system minimization****
minimisation for heated system****
&cntrl****
imin=1, maxcyc=1000, ncyc=500,****
igb=5, ntb=0,****
cut = 16, rgbmax = 16, saltcon = 0.2,****
ntpr=100****
/****
~****
~****
** **
*3. heat the system*
heating system from 0 K to 600K during 100 ps.****
&cntrl****
nstlim = 50000, dt = 0.002,****
ntt = 1, tautp = 1.0,****
tempi = 0, temp0 = 600, ntc =2, ntf = 2,****
ntpr =100, ntwx = 100,****
ntb = 0, igb = 5, ****
cut = 16, rgbmax = 16, saltcon = 0.2,****
nmropt = 1,****
/****
&wt****
type = 'TEMP0', istep1 = 0, istep2 =50000, value1 = 0, value2=600,****
/****
&wt type = 'END'****
/****
** **
*4**. **generate the full unfolded conformation*
** **
To generate the initial extend structure, a heating method was used to a
known NMR structure (PDB code:1ag2), enabling it to unfold at 600 K for 40
ns of LD simulation to result in an extended conformation.****
here, I do not sure whether I do the restart MD(set irest = 1, ntx = 5, or
irest = 0, ntx = 1), Simulation results is difference, which setting should
I use?
40nsld.inp****
enabling the heated NMR structure to unfold at 600 K for 40ns of LD
simulation****
&cntrl****
irest = 1, ntx = 5,****
nstlim = 20000000, dt = 0.002,****
ntt = 3, gamma_ln = 1.0,****
tempi = 600,temp0 = 600,****
ntb = 0, igb = 5,****
ntpr = 500, ntwx = 1000, ntwr = 2000000,****
ntc = 2, ntf = 2,****
cut = 16, rgbmax = 16, saltcon = 0.2,****
/
600kmd.inp****
enabling the heated NMR structure to unfold at 600 K for 40ns of LD
simulation****
&cntrl****
irest = 0, ntx = 1,****
nstlim = 20000000, dt = 0.002,****
ntt = 3, gamma_ln = 1.0,****
tempi = 600,temp0 = 600,****
ntb = 0, igb = 2,****
ntpr = 500, ntwx = 1000, ntwr = 2000000,****
ntc = 2, ntf = 2,****
cut = 16, rgbmax = 16, saltcon = 0.2,****
/
40nsld.out irest = 1, ntx = 5,****
600kmd.out irest = 0, ntx = 1****
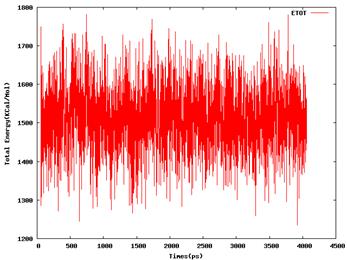
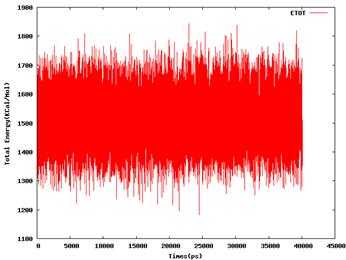
[image: 40nsld.etot.png]****
[image: 600kmd.etot.png]
** **
5. local minimization after heating system****
use the mdin file which is same to step 3 above。****
** **
6. equilibrate the every replica****
** **
equilibrate.mdin****
** **
equilibration 20 ns, every 10ps save output. <- Is this must to do
equilibrate for 20 ns?**
equilibration****
&cntrl****
irest=0, ntx=1,****
nstlim=10000000, dt=0.002,****
irest=0, ntt=3, gamma_ln=1.0,****
temp0=XXXXX, ig=RANDOM_NUMBER,****
ntc=2, ntf=2, nscm=1000,****
ntb=0, igb=5,****
cut = 16, rgbmax = 16, saltcon = 0.2,****
ntpr=5000, ntwx=5000, ntwr=10000000,****
nmropt=1,****
/****
&wt TYPE='END'****
/****
DISANG=system_chir.dat****
** **
7. REMD simulation****
remd 65ns exchange every 2ps <- Here I have been confused, how often to
exchange is more appropriate ?****
** **
remd.mdin****
remd 65ns exchange every 2ps****
&cntrl****
irest=0, ntx=1,****
nstlim=1000, dt=0.002,****
irest=0, ntt=3, gamma_ln=1.0,****
temp0=XXXXX, ig=RANDOM_NUMBER,****
ntc=2, ntf=2, nscm=1000,****
ntb=0, igb=5,****
cut = 16, rgbmax = 16, saltcon = 0.2,****
ntpr=100, ntwx=1000, ntwr=100000,****
nmropt=1,****
numexchg=32500,****
/
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: clip_image004.jpg)
(image/jpeg attachment: clip_image002.jpg)
