Date: Thu, 31 Jan 2013 17:42:42 +0100 (CET)
Hi Aneesh,
I gave it my best shot, but it is not exact since it is a smaller analogue than your molecule. Attached you will see the molecule I used, its partial atomic charges (via R.E.D. and 4 conformations for MEP generation), the atom types (using your molecule's assignment). I had to add three hydrogens to your force field due to the use of the analogue. The energy rotational curve is for C3-C1-S5-C23 rotation from 0 to 330 degrees. The QM10 is for constraint optimization at HF/6-31G(d) theory. The MM curve uses your force field, with the addition of the parameters needed for the added hydrogens - I "borrowed" their values from similar parameters within your force field. Perhaps you can check this torsion angle in your MD simulation, and it may match the minima seen within the MM curve. To do this better, it would take much more time. I hope this is of some help.
Cheers,
Karl
----- Original Message -----
From: "aneesh cna" <aneeshcna.gmail.com>
To: "AMBER Mailing List" <amber.ambermd.org>
Sent: Thursday, January 31, 2013 12:35:56 PM
Subject: Re: [AMBER] Fwd: Regarding the problem with ligand conformation
Hi Karl,
Thanks for you interest. I have used unique key words to manually control
the FF parameters, in way to ensure that I am using the best matching FF
parameter from the GAFF library. I have attached the PREP and FRCMOD file
for your reference.
Thanks and regards
Aneesh
On Thu, Jan 31, 2013 at 2:40 PM, Karl N. Kirschner <
kkirsch.scai.fraunhofer.de> wrote:
> Hello Aneesh,
>
> I have been thinking about your problem some, and I am curious about the
> performance of the parameters within your ligand. Could you send an image
> of your molecule with the atom type definition labeled next to the
> corresponding atom? (I assume you used parmchk after running antechamber to
> supply any missing parameters, correct?).
>
> Cheers,
> Karl
>
> ----- Original Message -----
> From: "aneesh cna" <aneeshcna.gmail.com>
> To: "AMBER Mailing List" <amber.ambermd.org>
> Sent: Wednesday, January 30, 2013 8:22:33 AM
> Subject: Re: [AMBER] Fwd: Regarding the problem with ligand conformation
>
> I maintain the system tepmerature at 300K. As Karl suggested, to see there
> is any effect of temperature on ligand conformation, I would like to try
> out few simulations possibly in the range of 283K - 293K.
>
> On Wed, Jan 30, 2013 at 11:31 AM, Bill Ross <ross.cgl.ucsf.edu> wrote:
>
> > > what
> > > could be the lowest possible temperature where I can do a classical MD
> > > simulation?
> >
> > What would be your ideal?
> >
>
>
> >
> > Bill
> >
> > aneesh cna <aneeshcna.gmail.com> wrote:
> >
> > > Hi Karl,
> > >
> > > Thanks for the reply. Since testing the fitness of each force-field
> terms
> > > will be time consuming, I fee it could be better to check the role of
> > > temperature on ligand conformation before getting into those QM-MM
> > > calculations. Looking from this aspect, I have a quick query that what
> > > could be the lowest possible temperature where I can do a classical MD
> > > simulation?
> > >
> > >
> > > Thanks in advance
> > >
> > >
> > > Regards,
> > > Aneesh
> > >
> > >
> > > On Tue, Jan 29, 2013 at 9:29 PM, Karl N. Kirschner <
> > > kkirsch.scai.fraunhofer.de> wrote:
> > >
> > > > Hi Aneesh,
> > > >
> > > > It is difficult to say if your simulations went wrong or not. For
> > one,
> > > > maybe your ligand is sampling conformations that are appropriate for
> a
> > room
> > > > temperature solution-phase model (assuming that is what you are
> > doing). The
> > > > crystal structure was likely solved at a much lower temperature,
> > resulting
> > > > in the preferred conformation you see in the PDB file.
> > > >
> > > > With that said, one could investigate how well the different
> > force-field
> > > > terms that you are using perform at reproducing known experimental
> > > > observables in analogues, if available. Alternatively you could see
> how
> > > > they reproduce QM-generated rotational curves for that molecule or a
> > > > smaller analogue. Doing so may provide you some confidence in their
> > use in
> > > > MD simulations. (Be forewarned, this can be time consuming to
> > accomplish.)
> > > >
> > > > Best regards,
> > > > Karl
> > > >
> > > > ----- Original Message -----
> > > > From: "aneesh cna" <aneeshcna.gmail.com>
> > > > To: "AMBER Mailing List" <amber.ambermd.org>
> > > > Sent: Tuesday, January 29, 2013 2:12:38 PM
> > > > Subject: [AMBER] Fwd: Regarding the problem with ligand conformation
> > > >
> > > > Dear Amber users,
> > > >
> > > > I am working with the protein-ligand systems. The protein-ligand
> > complex
> > > > structure was obtained from Protein Data Bank. Before starting the
> > > > simulation, I have carried out QM calculation ( b3lyp/6-311+g* ) of
> > ligand
> > > > to get the optimized structure and ESP fitted point charge for the
> > atoms.
> > > > The optimized ligand geometry was similar to the crystal structure.
> > Then, I
> > > > have used the antechamber module of Amber to prepare the parameter
> > file (
> > > > PREP and FRCMOD files) for the ligand, where I used the GAFF force
> > field.
> > > >
> > > > Unfortunately, the ligand geometry has been changed drastically
> during
> > the
> > > > initial stages of production run ( after ~1ns of equilibration phase)
> > of
> > > > simulation. The ligand keep its proper conformation ( i.e. close to
> > > > crystal conformation) for the initial ~1 ns phase of equilibration.
> Is
> > it a
> > > > problem with force field? . If so ( i.e., if there is any problem
> with
> > > > ligand FF parameters), the observed conformational changes should
> > happen in
> > > > the initial stage of the simulation rather than at the end of ~1ns
> > > > equilibration.
> > > >
> > > >
> > > > Can anyone help me to figure out what went wrong with my simulation.
> > > >
> > > >
> > > > I have attached three snapshot of the ligand (crystal structure, QM
> > > > optimized, after equilibration respectively), for your reference.
> > > >
> > > >
> > > >
> > > > Thanks in advance
> > > >
> > > >
> > > > Sincerely
> > > > Aneesh
> > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > > _______________________________________________
> > > > AMBER mailing list
> > > > AMBER.ambermd.org
> > > > http://lists.ambermd.org/mailman/listinfo/amber
> > > >
> > > _______________________________________________
> > > AMBER mailing list
> > > AMBER.ambermd.org
> > > http://lists.ambermd.org/mailman/listinfo/amber
> >
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
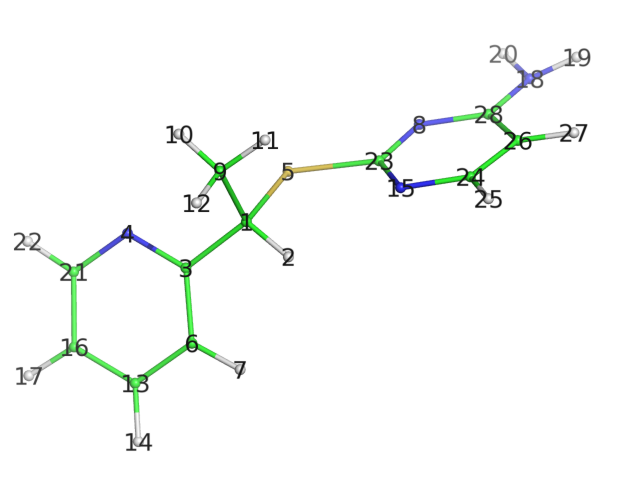
(image/png attachment: atom_labels.png)
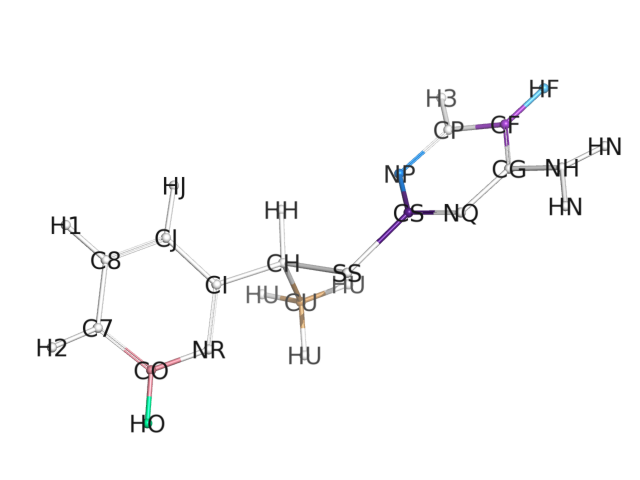
(image/png attachment: atom_types.png)
(image/png attachment: rotation_QM.png)

(image/png attachment: Energy_curves.png)
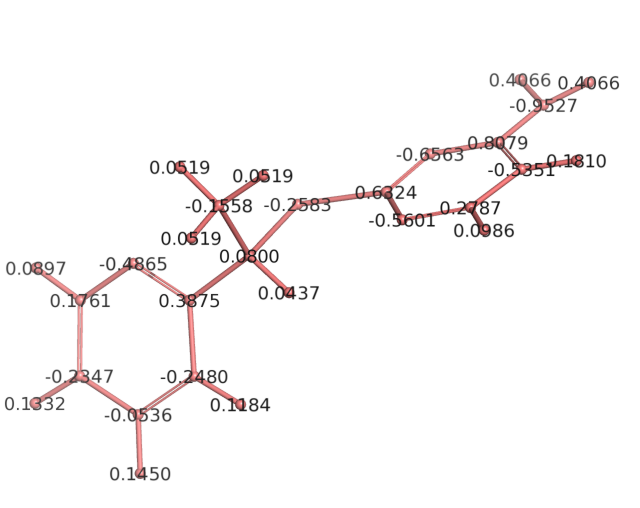
(image/png attachment: partial_atomic_charges.png)
