Dear Amber community
I got an output Gaussian file (in Gaussian03 windows version) using the
command “#HF/6-31G* SCF=tight Test Pop=MK iop(6/33=2) iop(6/42=6) opt”.
This was converted to .ac file format in antechamber and did RESP charge
fitting subsequently.
But I have two queries.
(1) I couldn’t find out how to generate a Gaussian file within an *
antechamber*.
(2) I downloaded the following file from internet and found that they have
generated a mol2 file in Step 1 and subsequently generated Gaussian input
files using antechamber.
But how do they generate the mol2 files containing both coordinates and
charges for each atom using Gaussian 03W?
Best regards,
Debolina Mitra
http://ambermd.org/antechamber/pro4.html
4-Hydroxyl-Proline (PR4)
- Step 1:
Prepare two mol2 files: alpha <http://ambermd.org/antechamber/pro4a.mol2>and
beta <
http://ambermd.org/antechamber/pro4b.mol2>conformatons for
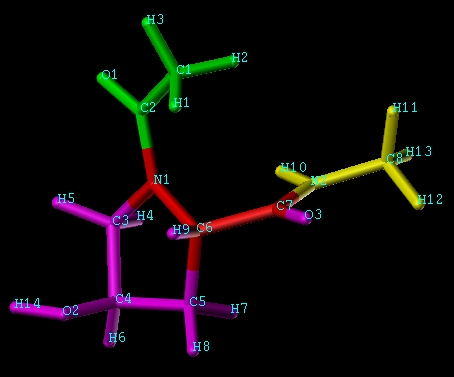
ACE-PR4-ACE (Take a look at the molecular structure
<
http://ambermd.org/antechamber/pro4.jpg>)
- Step 2:
Generate two gaussian input files: alpha
<
http://ambermd.org/antechamber/pro4a.gau>and beta
<
http://ambermd.org/antechamber/pro4b.gau>using the following
antechamber command:
*antechamber -fi mol2 -fo gzmat -i pro4a.mol2 -o pro4a.gau
antechamber -fi mol2 -fo gzmat -i pro4b.mol2 -o pro4b.gau
Do the following modifications on pro4a.gau and pro4b.gau:
change opt to popt in the keyword section
freeze t19 and t21 during the minimization with a tag of "F" *
- Step 3:
Run gaussian to get two output files: alpha
<
http://ambermd.org/antechamber/pro4a.out>and beta
<
http://ambermd.org/antechamber/pro4b.out>
- Step 4:
Extract ESP files from gaussian output files ( alpha
<
http://ambermd.org/antechamber/pro4a.esp>and beta
<
http://ambermd.org/antechamber/pro4b.esp>) and merge them together pro4.esp
<
http://ambermd.org/antechamber/pro4.esp>; then prepare input files for
two-stage resp fitting ( Step I
<
http://ambermd.org/antechamber/pro4-step1.respin>and Stage II
<
http://ambermd.org/antechamber/pro4-step2.respin>); finally run resp
program to get resp charge ( Pro4.crg
<
http://ambermd.org/antechamber/pro4.crg>).
*
espgen -i pro4a.out -o pro4a.esp
espgen -i pro4b.out -o pro4b.esp
cat pro4a.esp pro4b.esp >pro4.esp
respgen -i pro4a.mol2 -o pro4-step1.respin -f resp1 respgen -i
pro4a.mol2 -o pro4-step2.respin -f resp2
Manually revision pro4-step1.respin and pro4-step2.respin are necessary
in this example since respgen cannot generate input files for multiple
molecules. *
- Step 5:
Load pro4.crg and generate an ac file ( pro4.ac
<
http://ambermd.org/antechamber/pro4.ac>).
*antechamber -fi mol2 -fo ac -i pro4a.mol2 -o pro4.ac -c rc -cf pro4.crg
However, if one only use one conformation, he may generate an ac file
from the gaussian output file with charge method tag set to "resp"
directly. *
- Step 6:
Read in pro4.ac and a mainchain defination file mainchain.pro4
<
http://ambermd.org/antechamber/mainchain.pro4>to generate prep input
file 4-OH-PRO <
http://ambermd.org/antechamber/pro4.prepi>for 4-OH-PRO.
*In mainchain defination file, HEAD_NAME is the atom connected to the
immediately previous residue and TAIL_NAME is the atom connected to the
immediately behide reside; OMIT_NAME(s) is (are) not a part of the residue,
however they are necessary to maintain a proper chemical environment;
MAINCHAIN are mainchain atoms (HEAD_NAME and TAIL_NAME are mainchain atoms
automatically); PRE_HEAD_TYPE and POST_TAIL_TYPE N are the atom types of
atoms in other residues that are connected to the head and tail,
respectively. CHARGE is the net charge of the residue in question.
prepgen -i pro4.ac -o pro4.prepi -f prepi -m mainchain.pro4 -rn PR4 -rf
pro4.res
For PR4 <
http://ambermd.org/antechamber/pro4.jpg>, HEAD_NAME is N1;
TAIL_NAME is C7 and all the atoms in green and yellow are omitted atoms.
The PRO_HEAD_TYPE and POST_TAIL_TYPE are C and N respectively *
- Step 7:
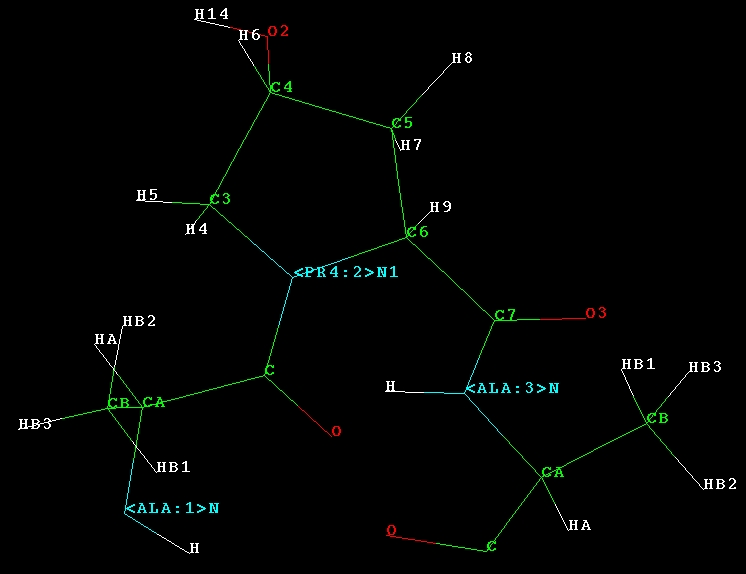
With pro4.prepi, one may use leap to generate topolgy files for sander
etc. Here we generate a topology file
<
http://ambermd.org/antechamber/ala-pr4-ala.prmtop>for ALA-PRO4-ALA with
leap <
http://ambermd.org/antechamber/ala-pr4-ala.jpg>.
*loadamberprep pro4.prep
aa =sequence {ALA PR4 ALA}
saveamberparm aa ala-pr4-ala.prmtop ala-pr4-ala.prmcrd *
--
Dr. Debolina Mitra
Assistant Professor
RBC College, Naihati
North 24-Parganas
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Thu May 24 2012 - 02:00:03 PDT
{kind=link}
{kind=link}
