Date: Tue, 28 Feb 2012 17:16:04 +0800
Hi Ben,
It took such a long time for the parameter validation of 3H68,I could
give the final results to you follow.Before I performed the MMPBSA
calculation,I have performed the below MD simulation: heat 500ps;density
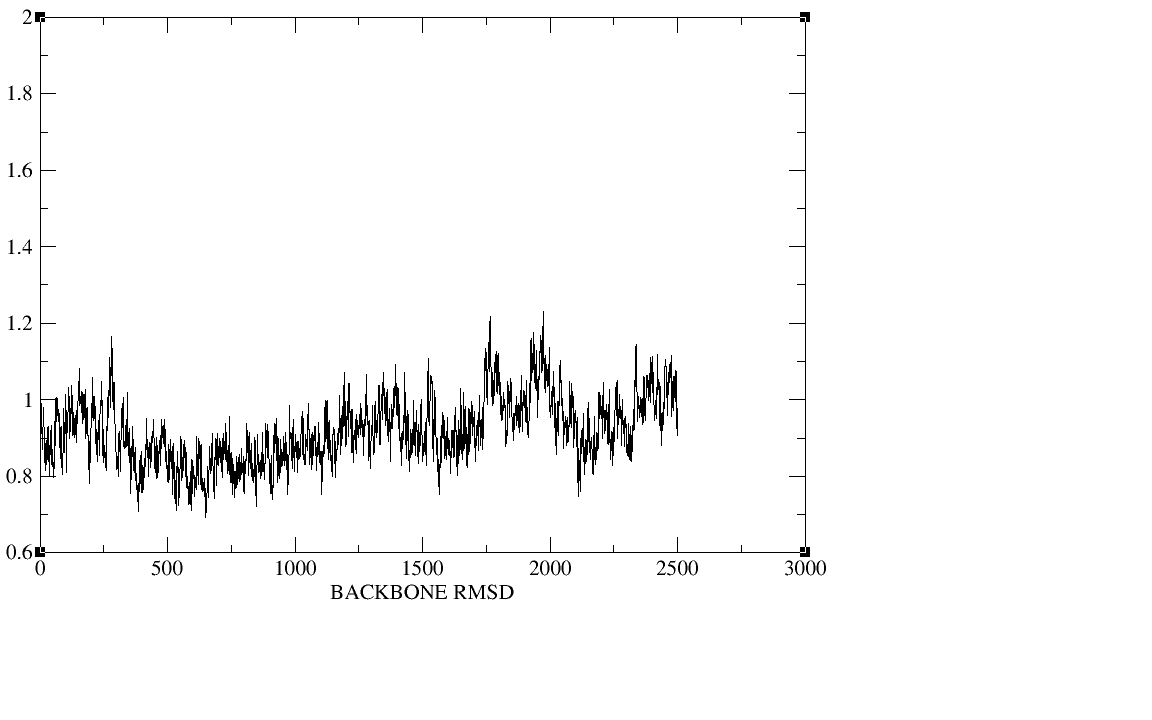
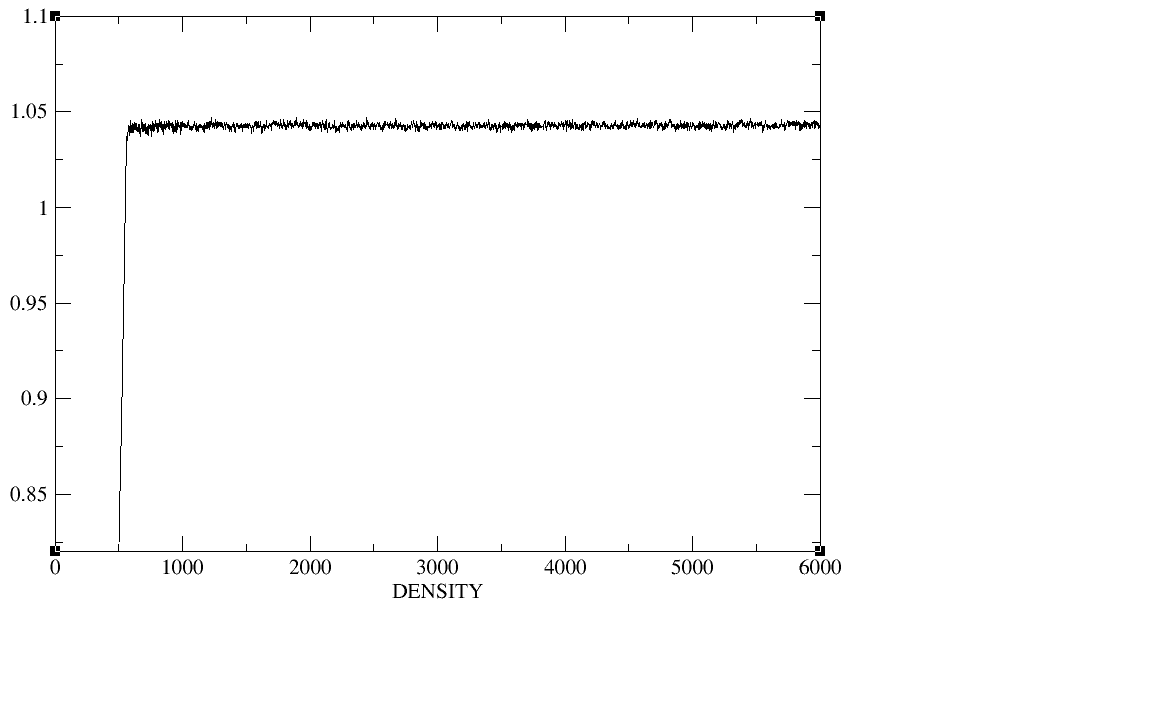
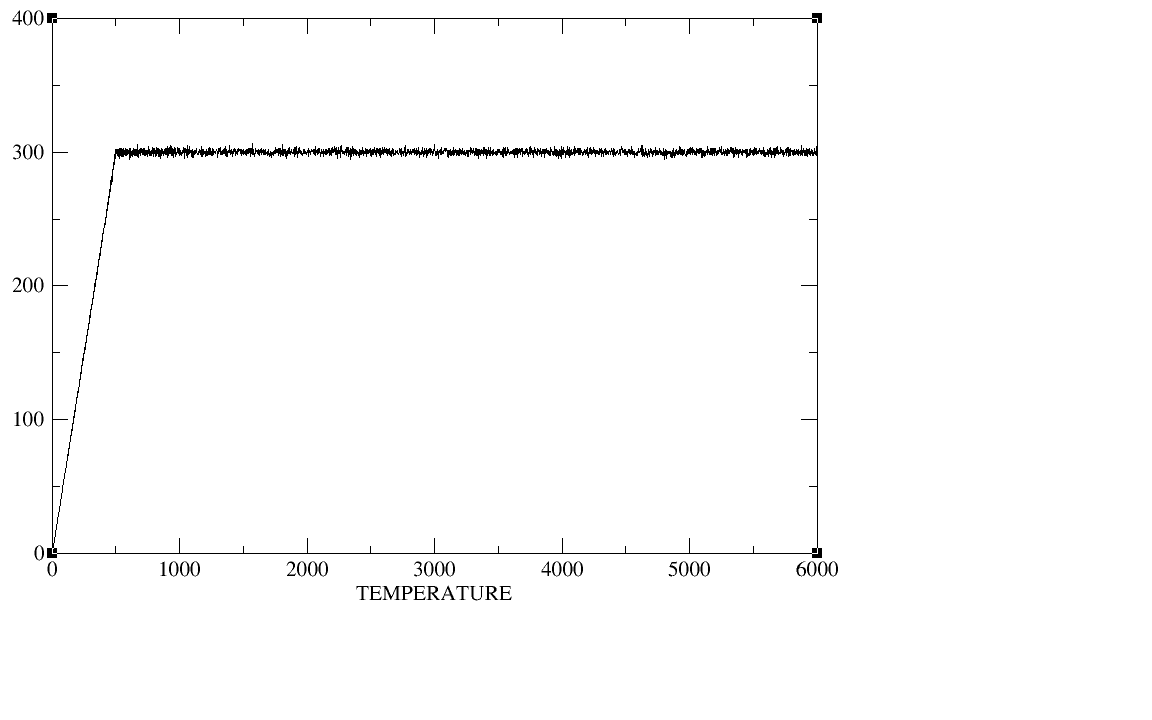
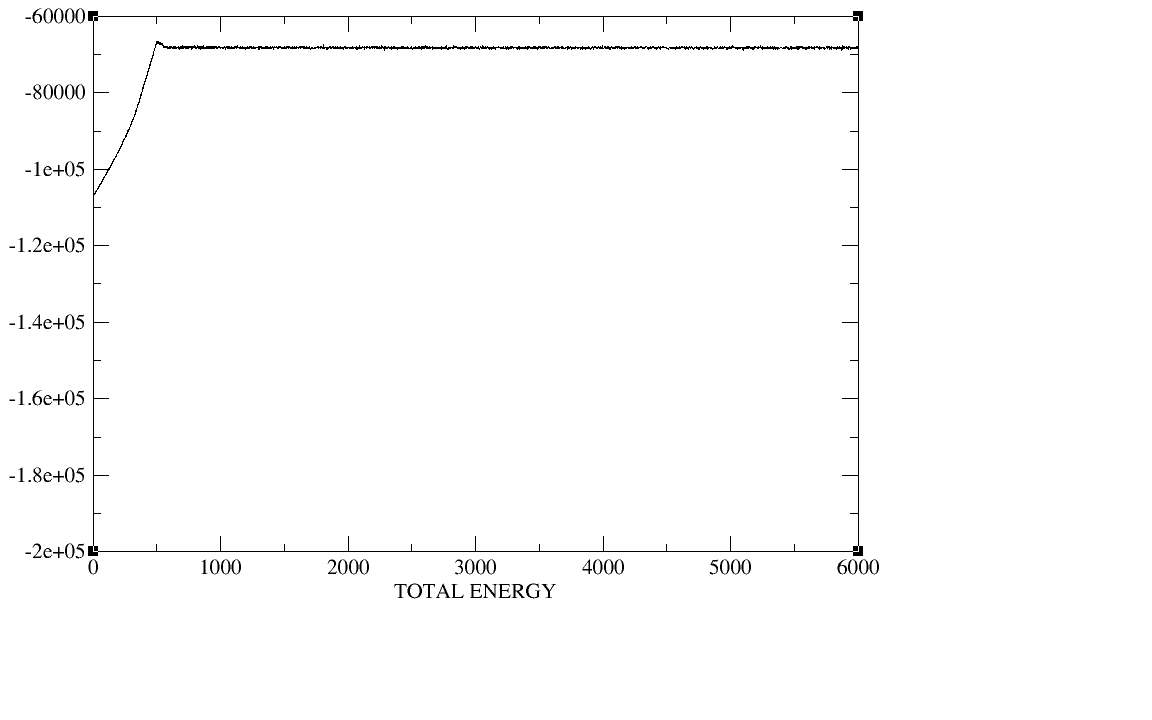
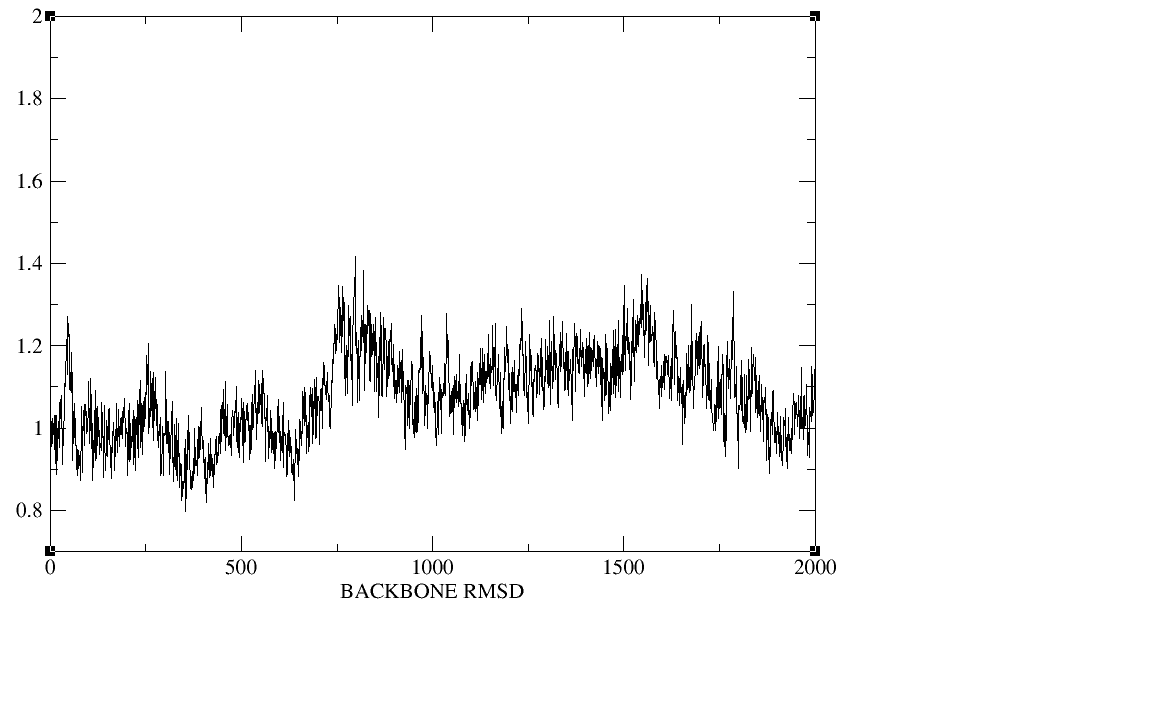
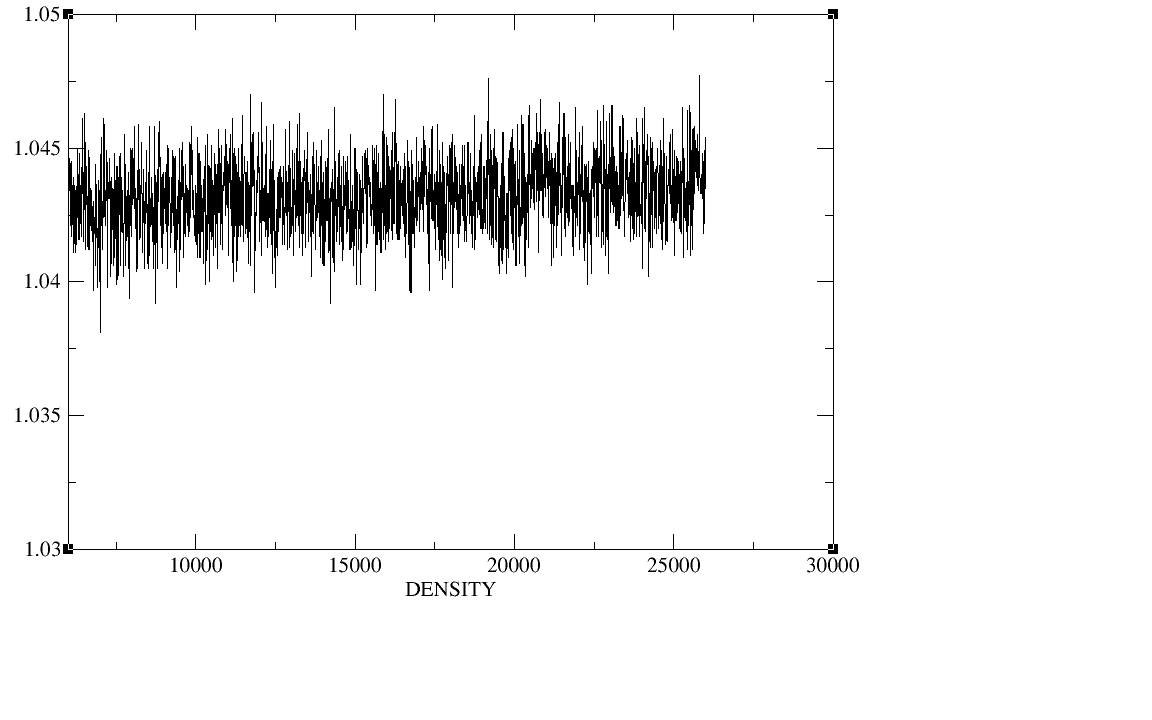
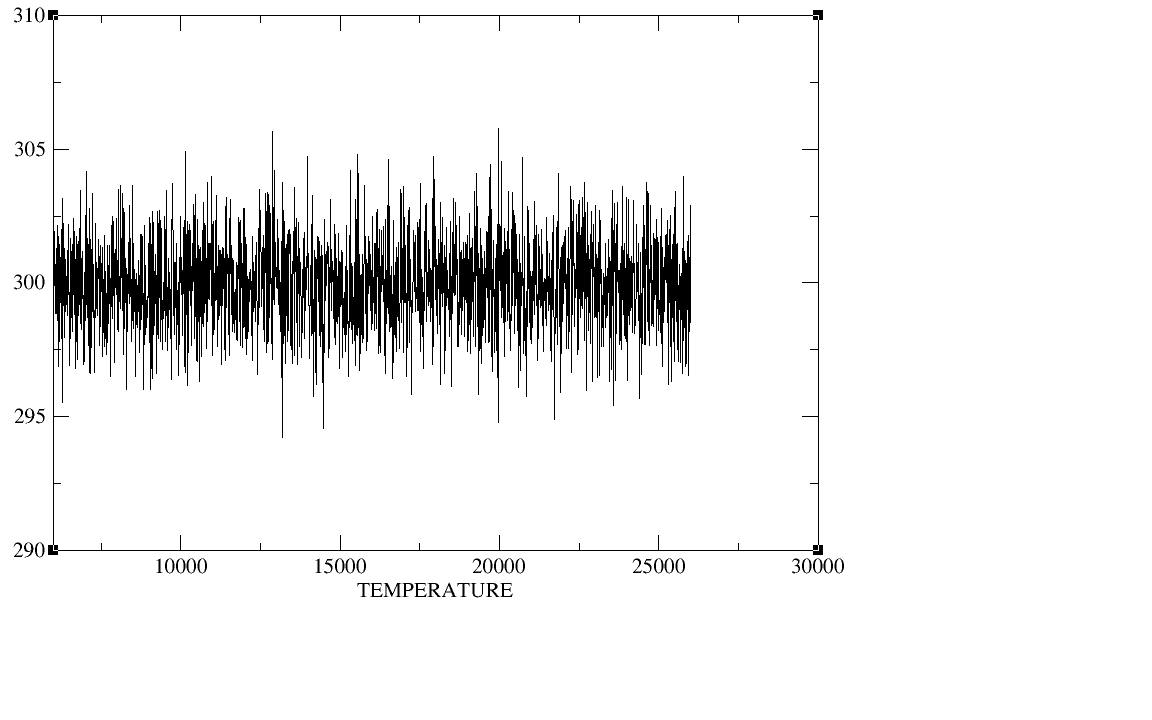
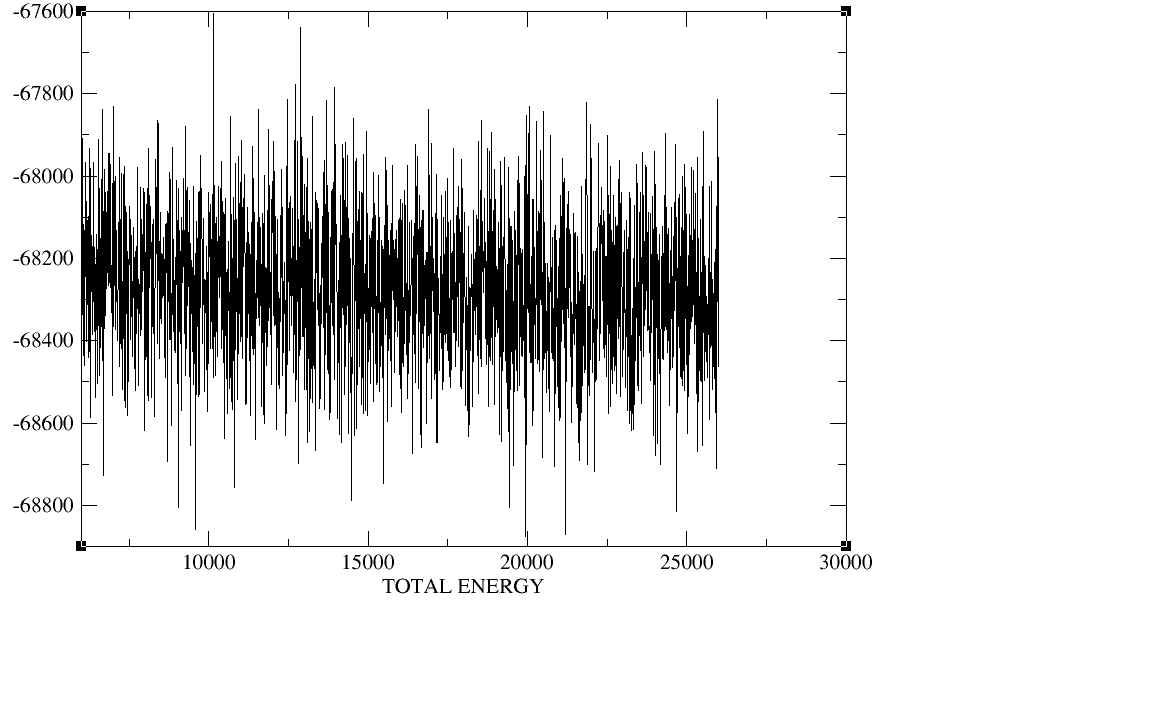
500ps;equil 5ns;production 20ns.I checked the plot of the density,
temperature, total energy and backbone RMSD during the heat,the
density,and the equilibration,The density, temperature,total energy,and
backbone RMSD plots have all clearly converged by the end of our
equilibration period.In production period those plots as well as the
three in the above(I send all the plots in the attachment) .I used VMD
to save out random conformations of the complex by reading over 26ns MD
trajactory,and superimposed these conformations to the crystal structure
named 3H68 by pymol,I found that the small molecule,two ZN2+ ions,and
the around amino acids which coordinates two ZN2+ were so stable with
the RMSD value 0.676~0.845 compared crystal structure 3H68.
I encountered a new problem about the small molecule whose comformation
have been a bit distorsion in its framework that included oxygen
bridge,but the structure of the small molecule was still intact,not be
deformed as before used the wrong charge.This distorsion like a cycle
around equil and production period,some frames is well and some is
distorted.I also sent one of the pdb of conformations with the small
molecule(prod1-ca1.pdb) that be distorted and a initial small molecule
pdb(ca.pdb) to you.Could the random distorsion for the small molecule
conformation in all of the MD period belong to a normal situation?Please
give some advises.
Best Regards
JiYuan
> Hi JiYuan,
>
> My apologies for the slow response time.
>
> On 4/01/2012, at 3:07 AM, JiYuan Liu wrote:
>
>> I used the command "antechamber -i ca.out -fi gout -o ca.prepin -fo prepi -c resp -s 2 *-nc -2*" to rebuild ca.prepin of my ligand,I used another command "prep2xml" to build the new calib.xml for my ligand,at this moment I encountered a new small problem that it represented excessed symbol	 in the lines of the new calib.xml.I have attached the log and output files after performed "prep2xml",Please have a look for them.
> I haven't seen this problem before. It looks as though it may be a text encoding issue. Do you know what text encoding you were using at the time you prepared your input? Martin may be able to advise better on this, but I suspect MCPB probably works best with ASCII or UTF-8 encoded text.
>
>> Although I found the small problem in the above,I just delete the symbol "	" in the calib.xml and replaced the old calib.xml.Everything looked fine when I performed "sh run.MCPB.csh",I found no difference at 3H68_OH_sidechain_opt.com and 3H68_OH_large_mk.com as before I used the old wrong calib.xml,so I didn't recalculate the Side Chain Model Optimization/Frequency Calculation and Large Model Charge Calculation.
> Good. I'm pleased that you could save time in that way.
>
>> Then I performed 5000 steps minimization and 10ps molecule dynamics,the small molecule,two ZN2+ ions,and the around amino acids which coordinates two ZN2+ were so stable.I need your help to give me a judge whether my this solution is reasonable.
> We can't really offer a parameter validation service. Ultimately, you'll need to be the judge. Do the parameters seem reasonable in themselves? When you simulate with them, do the results make physical and chemical sense?
>
> Best,
> Ben
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Jiyuan Liu Key Laboratory of Plant Protection Resources and Pest Management,National Ministry of Education Northwest A&F University Yangling, Shaanxi China 712100 Phone: 86-29-87092190
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: equil-backbone_rmsd.png)
(image/png attachment: equil-density.png)
(image/png attachment: equil-temp.png)
(image/png attachment: equil-total_energy.png)
(image/png attachment: prod-backbone_rmsd.png)
(image/png attachment: prod-density.png)
(image/png attachment: prod-temp.png)
(image/png attachment: prod-total_energy.png)
- application/vnd.palm attachment: prod1-ca1.pdb
- application/vnd.palm attachment: ca.pdb
