Date: Tue, 27 Sep 2011 19:44:14 +0100
Dear Amber developers,
I try to resend my email that probably was not read/received.
Any hint to fix my RMSD values would be really appreciated.
Cheers,
M
---------- Messaggio inoltrato ----------
Da: Massimiliano Porrini <M.Porrini.ed.ac.uk>
Date: 26 settembre 2011 18:18
Oggetto: Re: [AMBER] fixing rmsd values
A: AMBER Mailing List <amber.ambermd.org>
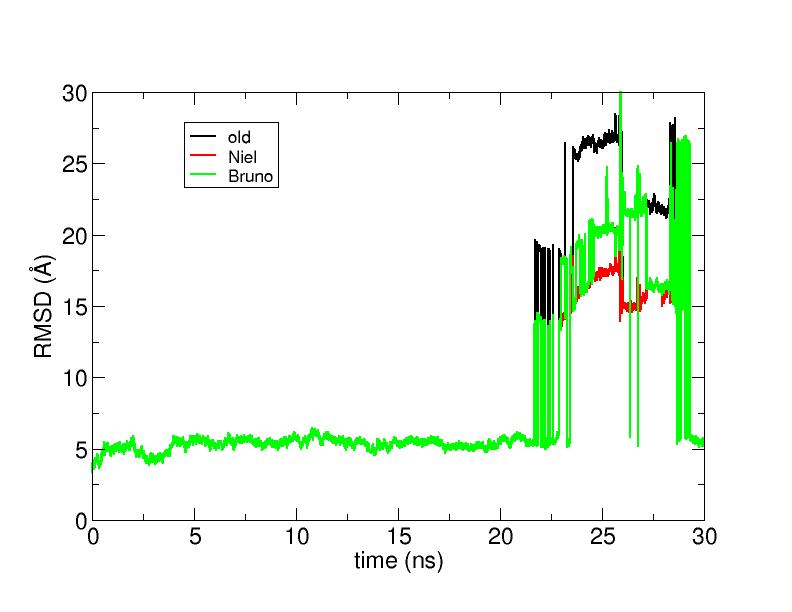
I really do not want to annoy you with this subject again,
but even with the commands suggested by Bruno Rodrigues
it did not work (it got actually worse, as you can see
from the attached graph, I apologise about giving personal names
to the solid lines, but it was the best way to make it clear).
Besides now I am more confused than before about the right keywords to use
to obtain a nice RMSD time series.
I am very thankful for the hints though.
All the best,
Il 26 settembre 2011 16:54, Bruno Rodrigues <bbrodrigues.gmail.com> ha scritto:
> Try this one:
>
> trajin tetra_dyn.dcd
> center :1-11 mass
> image origin center
> center :1-22 mass
> image center familiar
> center :1-33 mass
> image center familiar
> center :1-44 mass
> image center familiar
> image familiar
> reference tetra.pdb
> rms reference mass out rmsd.dat .C,CA,N time 2
>
> On Mon, Sep 26, 2011 at 11:46 AM, Massimiliano Porrini
> <M.Porrini.ed.ac.uk>wrote:
>
>> Dear Niel and dear all,
>>
>> Thanks very much for the hint, but, unless I am missing something here,
>> unfortunately it did not work in my case.
>>
>> As I think my problem is exactly the same as the one presented in the post
>> suggested by Niel, I updated my script commands:
>>
>> trajin tetra_dyn.dcd
>> center :1-11 mass origin
>> image origin center familiar
>> center :1-22 mass origin
>> image origin center familiar
>> center :1-33 mass origin
>> image origin center familiar
>> center :1-44 mass origin
>> image origin center familiar
>> reference tetra.pdb
>> rms reference mass out rmsd.dat .C,CA,N time 2
>>
>> And as you can see from the attached graph the RMSD has slightly
>> improved, but still I get those jumps at the end.
>>
>> Any idea of what's happening here would be really appreciated.
>>
>> All the best,
>>
>>
>> Il 25 settembre 2011 16:47, Niel Henriksen <niel.henriksen.utah.edu> ha
>> scritto:
>> > The Amber mailing list archive is a great resource to search:
>> > http://archive.ambermd.org/
>> >
>> > It so happens that this issue has been discussed numerous times.
>> > It is an imaging issue ... this post explains what to do nicely:
>> > http://archive.ambermd.org/201103/0607.html
>> >
>> > If you search the archive there are lots of examples and a few
>> > different approaches to dealing with it.
>> >
>> > Good luck,
>> > --Niel
>> > ________________________________________
>> > From: Massimiliano Porrini [M.Porrini.ed.ac.uk]
>> > Sent: Sunday, September 25, 2011 9:22 AM
>> > To: AMBER Mailing List
>> > Subject: [AMBER] fixing rmsd values
>> >
>> > Dear all,
>> >
>> > I have worked out the rmsd time series for a 30 ns trajectory
>> > of my system, which is a tetramer of a 11-residues peptide
>> > in explicit water, using the following ptraj commands:
>> >
>> > trajin tetra_dyn.dcd
>> > center :1-44 mass origin
>> > image :1-44 origin center
>> > reference tetra.pdb
>> > rms reference mass out rmsd.dat .C,CA,N time 2
>> >
>> > As you can see from the attached graph the rmsd time series bears
>> > some "jumps".
>> > These are due to the fact that the center of mass of one or more monomers
>> > exceeds the boundaries of my cell (truncated octahedron) during the
>> > simulation and
>> > generate the high values of the RMSD.
>> >
>> > Does anybody know how I might fix the RMSD values via any ptraj commands?
>> > Any suggestion would be really appreciated.
>> >
>> > Best,
>> >
>> > --
>> > Dr Massimiliano Porrini
>> > Institute for Condensed Matter and Complex Systems
>> > School of Physics & Astronomy
>> > The University of Edinburgh
>> > James Clerk Maxwell Building
>> > The King's Buildings
>> > Mayfield Road
>> > Edinburgh EH9 3JZ
>> >
>> > Tel +44-(0)131-650-5229
>> >
>> > E-mails : M.Porrini.ed.ac.uk
>> > mozz76.gmail.com
>> > maxp.iesl.forth.gr
>> >
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>> >
>>
>>
>>
>> --
>> Dr Massimiliano Porrini
>> Institute for Condensed Matter and Complex Systems
>> School of Physics & Astronomy
>> The University of Edinburgh
>> James Clerk Maxwell Building
>> The King's Buildings
>> Mayfield Road
>> Edinburgh EH9 3JZ
>>
>> Tel +44-(0)131-650-5229
>>
>> E-mails : M.Porrini.ed.ac.uk
>> mozz76.gmail.com
>> maxp.iesl.forth.gr
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>>
>
>
> --
> --
> Bruno Barbosa Rodrigues
> PhD Student - Physics Department
> Universidade Federal de Minas Gerais - UFMG
> Belo Horizonte - Brazil
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- Dr Massimiliano Porrini Institute for Condensed Matter and Complex Systems School of Physics & Astronomy The University of Edinburgh James Clerk Maxwell Building The King's Buildings Mayfield Road Edinburgh EH9 3JZ Tel +44-(0)131-650-5229 E-mails : M.Porrini.ed.ac.uk mozz76.gmail.com maxp.iesl.forth.gr -- Dr Massimiliano Porrini Institute for Condensed Matter and Complex Systems School of Physics & Astronomy The University of Edinburgh James Clerk Maxwell Building The King's Buildings Mayfield Road Edinburgh EH9 3JZ Tel +44-(0)131-650-5229 E-mails : M.Porrini.ed.ac.uk mozz76.gmail.com maxp.iesl.forth.gr
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: rmsd.jpg)
