Date: Sun, 25 Sep 2011 16:22:13 +0100
Dear all,
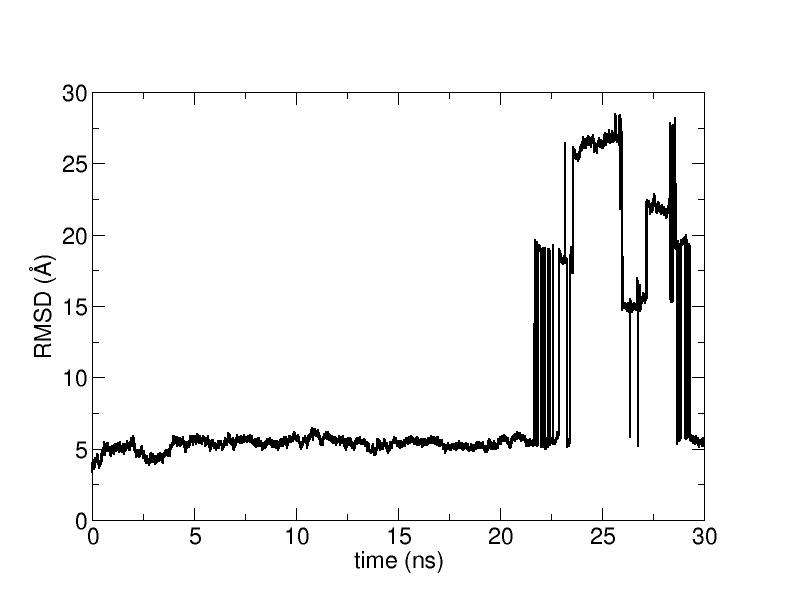
I have worked out the rmsd time series for a 30 ns trajectory
of my system, which is a tetramer of a 11-residues peptide
in explicit water, using the following ptraj commands:
trajin tetra_dyn.dcd
center :1-44 mass origin
image :1-44 origin center
reference tetra.pdb
rms reference mass out rmsd.dat .C,CA,N time 2
As you can see from the attached graph the rmsd time series bears
some "jumps".
These are due to the fact that the center of mass of one or more monomers
exceeds the boundaries of my cell (truncated octahedron) during the
simulation and
generate the high values of the RMSD.
Does anybody know how I might fix the RMSD values via any ptraj commands?
Any suggestion would be really appreciated.
Best,
-- Dr Massimiliano Porrini Institute for Condensed Matter and Complex Systems School of Physics & Astronomy The University of Edinburgh James Clerk Maxwell Building The King's Buildings Mayfield Road Edinburgh EH9 3JZ Tel +44-(0)131-650-5229 E-mails : M.Porrini.ed.ac.uk mozz76.gmail.com maxp.iesl.forth.gr
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: tetra_rmsd.jpg)
