Date: Tue, 16 Aug 2011 17:22:40 -0300
Hi Marc,
I've got the following error on the output of the simulation
| ERROR: Could not read coords from
/dados/bbr/bmsp/tip3pf/slow_heat/1D20_wat_tip3pf.heat19
On 8/16/11, Marc van der Kamp <marcvanderkamp.gmail.com> wrote:
> Bruno, the question is: do you have ****** instead of a floating point in
> the restart file that didn't work?
> If so, the coordinate value probably exceeded 1000 (or actually
> 999.9999999), the maximum possible in the .rst format, and would be the
> reason AMBER can't read it and continue simulation with it. That's the
> reason to use iwrap=1.
I got the following lines:
64.6583766 49.3786312 91.2950869 65.6254003 48.2428823 91.5518450
-176.0237426************ 358.1298288-175.2878018************ 357.9544071
-176.4867711************ 358.8610230 -48.4805095 112.0249683-104.5185947
-47.7891838 112.6572427-104.3223049 -48.6672397 111.6057582-103.6785790
So this is the reason for the error on the output.
> I'm not entirely sure if you tried re-running the step that created the
> problematic .rst file with iwrap=1 or not.
I didn't. I'm doing it right now, but I will have to set it up for the
same simulation time, otherwise I may not observe if the problem is
solved. And it will take at least 24 hours if I'm lucky to get 40
processors or more.
> Apart from whether coordinates in output files are imaged or not, I don't
> think there is a difference in running with iwrap=1 or iwrap=0 in
> sander/pmemd (although I could be wrong...). This means that there probably
> won't be a problem in switching to iwrap=1 in the step that generated the
> problematic .rst file.
>
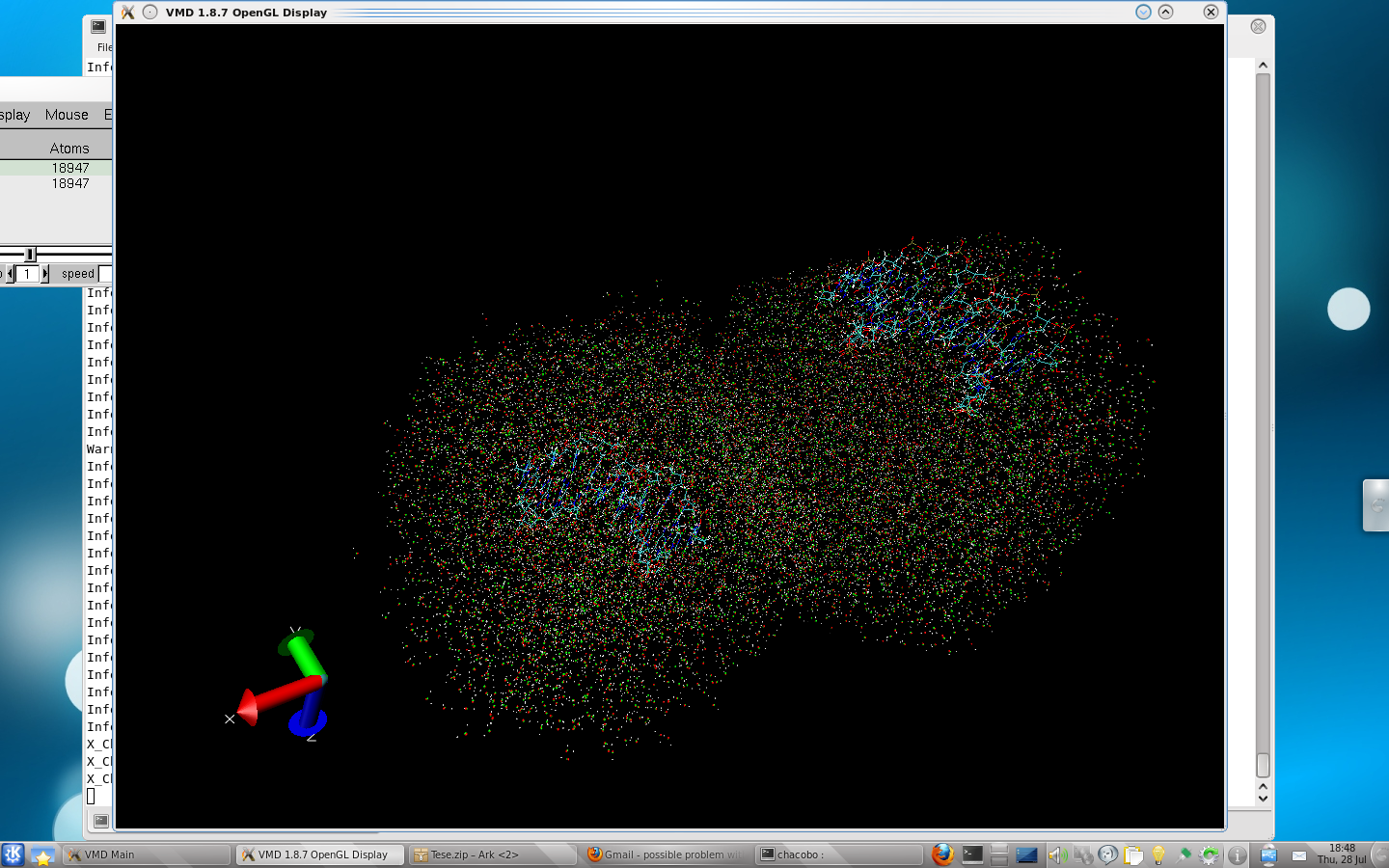
Enclosed is a VMD snapshot from 2 simulations, the one with iwrap = 1
represents the Right, while iwrap = 0 is the left one. You can see the
shift on the solute, as well as the breaking on some hydrogen bonds.
> --Marc
>
> On 16 August 2011 12:40, Bruno Rodrigues <bbrodrigues.gmail.com> wrote:
>
>> Well,
>>
>> Maybe I did not properly explian my problem:
>>
>> I realized that with iwrap=1 the DNA shifts to the border of the box, and
>> sometimes breaks the two strands apart. I reported the problem before at
>> the
>> list, but didn't get any clue.
>>
>> So I decided to switch iwrap off. As the simulation lasts for 40ns with a
>> T-jump from 300 up to 400K, I *guess* the problem is related with
>> molecules
>> very far from the solute. I read something about it on the developer's
>> list.
>> I know these molecules far from the solute are not a true problem, as I
>> can
>> always bring them back to the 1st box with ptraj.
>>
>> So here is the puzzle:
>>
>> I have the coordinate and restart files from the last simulation. The
>> restart generated does not work for the next step. The output complains
>> that
>> can not read the file. Then, I re-imaged the mdcrd files back to the first
>> cell. I would like to generate a restart file from the last frame of
>> *this*
>> mdcrd, as the molecules look fine in the first cell.
>>
>> But as you mentioned, there's no velocity information. Should I
>> equilibrate
>> it for longer before continue with the heating? What can I do to avoid it?
>> And did someone else have problems with iwrap?
>>
>> Thank you in advance.
>>
>> On Tue, Aug 16, 2011 at 8:26 AM, Marc van der Kamp <
>> marcvanderkamp.gmail.com
>> > wrote:
>>
>> > Perhaps to clarify:
>> > It appears that Bruno's problem is restarting a simulation, so a restart
>> > file generated as Jason suggested (i.e. without velocities) won't help
>> > in
>> > that case.
>> > What Jason does implicitly mention, is that Bruno probably doesn't have
>> > a
>> > problem, as long as he switches to iwrap=1 in the simulation. Even
>> > though
>> > the coordinates extracted from it may look weird, they are probably ok
>> > in
>> > the real periodic system.
>> > As Bruno himself already mentioned, you can process the .mdcrd / restart
>> > files later in ptraj to get coordinates that do look ok.
>> > So, as long as your energies etc. look fine, you can just continue MD
>> from
>> > the restart file generated with iwrap=1.
>> >
>> > --Marc
>> >
>> > On 16 August 2011 12:16, Jason Swails <jason.swails.gmail.com> wrote:
>> >
>> > > You can use cpptraj or ptraj to create restart files from the mdcrd:
>> > >
>> > > trajin my_traj.mdcrd 5 100 5
>> > > trajout my_snapshots.restrt restart
>> > >
>> > > However, note that you will *not* have any velocity information, so it
>> > can
>> > > never be a true restart. Therefore, you may be better off using the
>> > > restart
>> > > that has been working for you, but just looks weird.
>> > >
>> > > HTH,
>> > > Jason
>> > >
>> > > On Mon, Aug 15, 2011 at 10:32 PM, Bruno Rodrigues <
>> bbrodrigues.gmail.com
>> > > >wrote:
>> > >
>> > > > Dear all,
>> > > >
>> > > > I'm running a long simulation of DNA and water, with 20 different
>> > inputs.
>> > > > However, at the 19th, the simulation stopped, and did not restart.
>> > > >
>> > > > I searched on the list and found that, because I didn't use iwrap,
>> > maybe
>> > > > there are some coordinates very far from the origin and sander
>> > > > didn't
>> > > read
>> > > > it.
>> > > >
>> > > > However, if I use iwrap, it messes out the DNA inside the box. I
>> > managed
>> > > to
>> > > > bring the molecules back to the box by centering them. But how can I
>> > > > generate a restart file from the mdcrd ones?
>> > > >
>> > >
>> > > > Regards,
>> > > >
>> > > > --
>> > > > --
>> > > > Bruno Barbosa Rodrigues
>> > > > PhD Student - Physics Department
>> > > > Universidade Federal de Minas Gerais - UFMG
>> > > > Belo Horizonte - Brazil
>> > > > _______________________________________________
>> > > > AMBER mailing list
>> > > > AMBER.ambermd.org
>> > > > http://lists.ambermd.org/mailman/listinfo/amber
>> > > >
>> > >
>> > >
>> > >
>> > > --
>> > > Jason M. Swails
>> > > Quantum Theory Project,
>> > > University of Florida
>> > > Ph.D. Candidate
>> > > 352-392-4032
>> > > _______________________________________________
>> > > AMBER mailing list
>> > > AMBER.ambermd.org
>> > > http://lists.ambermd.org/mailman/listinfo/amber
>> > >
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org
>> > http://lists.ambermd.org/mailman/listinfo/amber
>> >
>>
>>
>>
>> --
>> --
>> Bruno Barbosa Rodrigues
>> PhD Student - Physics Department
>> Universidade Federal de Minas Gerais - UFMG
>> Belo Horizonte - Brazil
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
-- -- Bruno Barbosa Rodrigues PhD Student - Physics Department Universidade Federal de Minas Gerais - UFMG Belo Horizonte - Brazil
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: system.png)
