Date: Fri, 06 May 2011 12:30:05 +0200
Hi everybody,
I'm trying to work out the contribution of a buried water molecule to the Delta G of binding of a small organic molecule to a protein of 141 residues.
Based on earlier mailings to the list, I tried the following:
1. A test run to generate pdb files from my 2ns trajectory.
Used: ptraj my.prmtop closest.in
where <closest.in> was
trajin 2ns.mdcrd
trajout test.pdb pdb
closest 1 :1-142 first
The output was
1> CLOSESTWATERS: saving the 1 closest solvent molecules around atoms :1-142
The current solvent mask is :155-8445
PTRAJ: Successfully read in 200 sets and processed 200 sets.
The output was 200 pdb files of the complex with 13 Na+ atoms and one water molecule.
2. Generated the processed trajectory
Used: ptraj my.prmtop process.in
where <process.in> was
trajin 2ns.mdcrd
trajout 2nsHOH.mdcrd
closest 1 :1-142 first
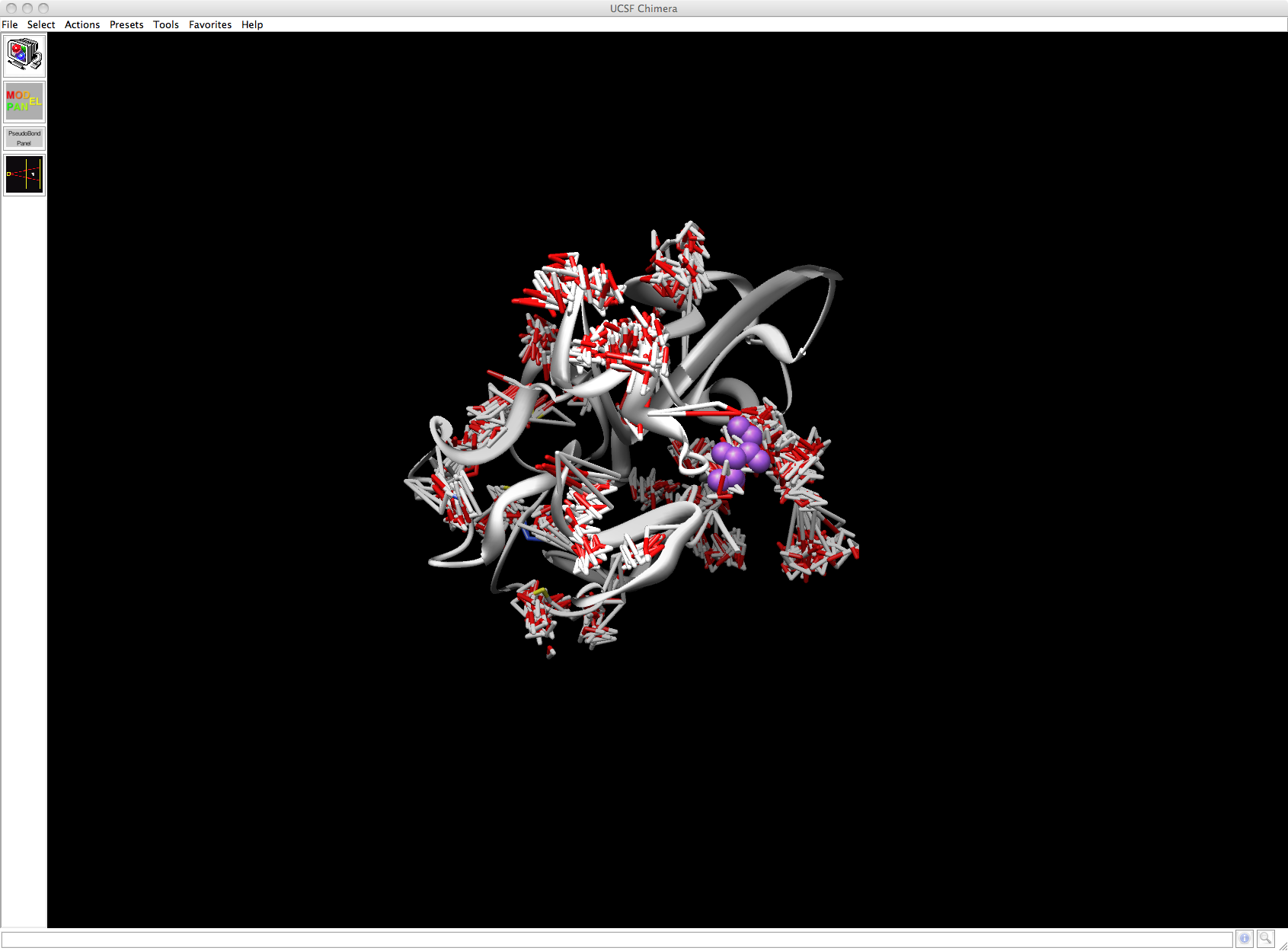
The visualisation of the 2nsHOH.mdcrd output is rather weird. I'm attaching a snapshot
It shows the closest HOH molecules clustered around the complex and the Na+ ions localised as a cluster around a specific part of the complex.
I'm pretty sure this is not what I'm supposed to deal with. So there's something wrong with the above syntax.
Could anyone please offer some suggestions.
You're help is much appreciated.
And a second related question.
MMPBSA.py will require a new topology file (protein/ligand/water). To generate this topology file one would need a starting pdb. Can one use any pdb of the 200 generated in step 1 above?
Thanks again
George
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
