Date: Fri, 08 Apr 2011 23:11:43 +0200
Hi everybody,
I'm trying to generate ligand input files for an MD ligand/protein simulation.
My ligand is caryophyllene (see snapshot)
The ligand caryo.pdb has been generated with Chimera and the caryo.mol2 file with antechamber.
Checking the unit ("check CARYO) gives the following output
Checking 'CARYO'....
Checking parameters for unit 'CARYO'.
Checking for bond parameters.
Checking for angle parameters.
Could not find angle parameter: cy - c2 - c3
Could not find angle parameter: c2 - cy - hc
There are missing parameters.
Unit is OK.
I'm reluctant to continue given that the above parameters are missing.
I'd appreciate any suggestion on solving this problem
A good evening to all
George
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
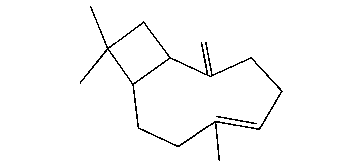
(image/gif attachment: beta-caryophyllene.gif)
