Date: Thu, 21 Oct 2010 11:13:45 -0700
Quoting Bill Ross <ross.cgl.ucsf.EDU>:
>> I tried to make bigger graphite about 200 atoms.
>> I just found that UCSF chimera can make this structure from the unit
>> cell pdb(2c.pdb) file by supercell operation and it can also generate
>> proper bonds between carbons and finally I can see a good graphite
>> with all 6-membered rings(c10-1.pdb).
>>
>> I loaded this pdb with previous generated prepi file from 2-carbon
>> unit cell(2c.prepi).
>> --------------------------------------------------
>> source leaprc.ff99SB
>> source leaprc.gaff
>> loadamberprep c2.prepi
>> gr=loadpdb c10-1.pdb
>> savepdb gr c10amb.pdb
>> solvatebox gr TIP3PBOX {0 0 20}
>> saveamberparm gr c10.prmtop c10.inpcrd
>> ---------------------------------------------------
>> After loading prmtop and inpcrd files, I see additional bonds between
>> two edge carbons. And after minimization with waters, I see tube like
>> structure instead of flat surface. I think leap generate wrong bonds.
>
> The most reliable way to see if leap adds any bonds is to look
> in xleap ("edit gr"). Also VMD should show this if you load
> prmtop.
>
I checked all the bonds in xleap, xleap simply ignored the bonds
connections from the input pdb files and makes really long bonds
between upper edge and lower edge.
>> ======================================
>> sam wat heat 1
>> &cntrl
>> imin = 0,
>> irest = 0,
>> ntx = 1,
>> ntb = 1,
>> cut = 10.0,
>> ntr = 1,
>> ntc = 2,
>> ntf = 2,
>> tempi = 0.0,
>> temp0 = 50.0,
>> ntt = 3,
>> gamma_ln = 1.0,
>> nstlim = 10000, dt = 0.0005
>> ntpr = 50, ntwx = 100, ntwr = 1000
>> /
>> Hold the SAM fixed
>> 50.0
>> RES 1 100
>> END
>> END
>> ===================================
>> And with the slow heating from 0 to 50K, I see NaN error again.
>> Could you give me any idea how to solve all these problem?
>
> That's fast heating to 50K, but such a low temp should be ok.
> (For slow heating, one would use a graduated temp restraint.)
> How many steps before the NaN error? I would set ntwx=1 to dump
> coordinates for every frame, then watch the movie (e.g. in VMD)
> and see what is happening.
>
> Bill
>
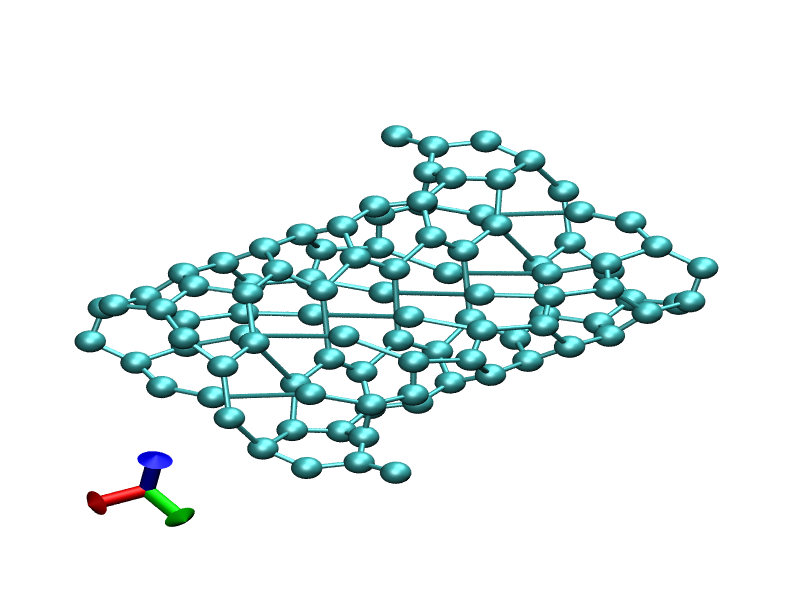
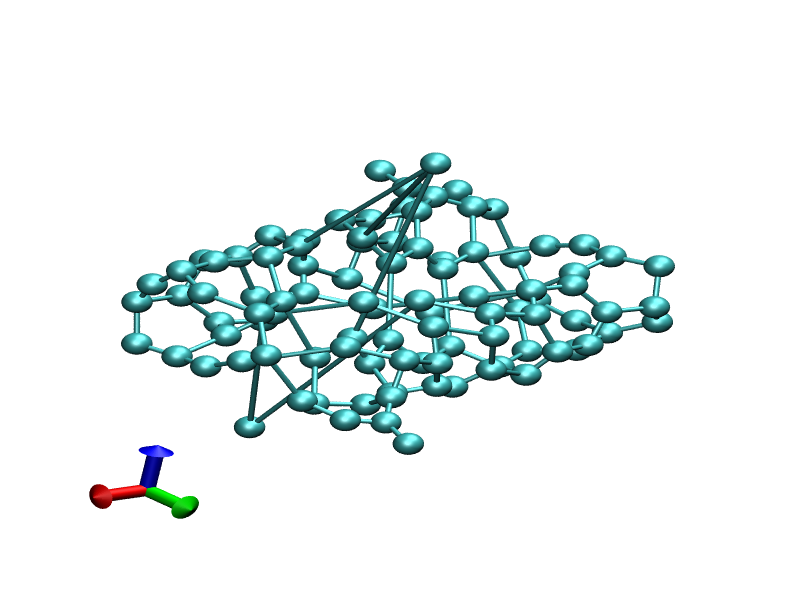
I changed ntt=2 and ntpr and ntwx=1. It starts with zigzaged tube like
structure(init.png) and around after 600 frame, I can see a jumped
atom with relatively long bond distance(final.png). I attached prmtop,
inpcrd and two other minimization rst files togeter.
Thank you.
Bongkeun Kim
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- text/plain attachment: c7.prmtop
- text/plain attachment: c7.inpcrd
- text/plain attachment: c7_min1.rst
- text/plain attachment: c7_min2.rst
(image/png attachment: init.png)
(image/png attachment: final.png)
