From: hirdesh kumar <hirdeshs8.gmail.com>
Date: Tue, 28 Sep 2010 15:06:44 +0530
Hi,
I am not able to check for the S-S bond as I can't distinguish various
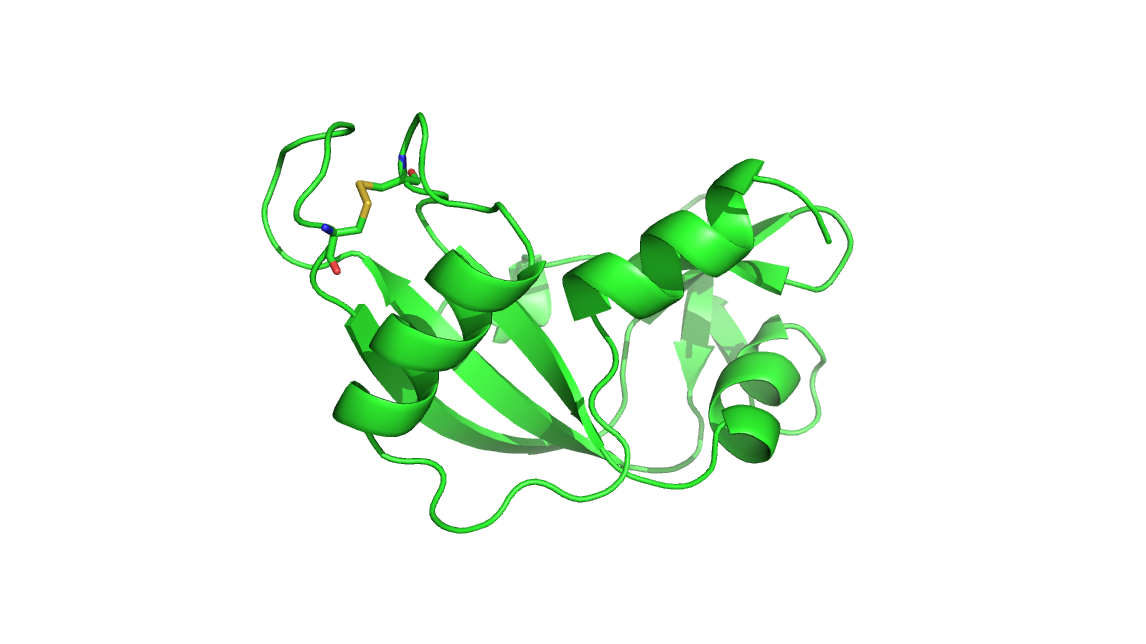
atoms for the residues in .parm7 file. I think the S-S bond was defined in
the beginning of minimization (as the corresponding ambpdb prepared .pdb
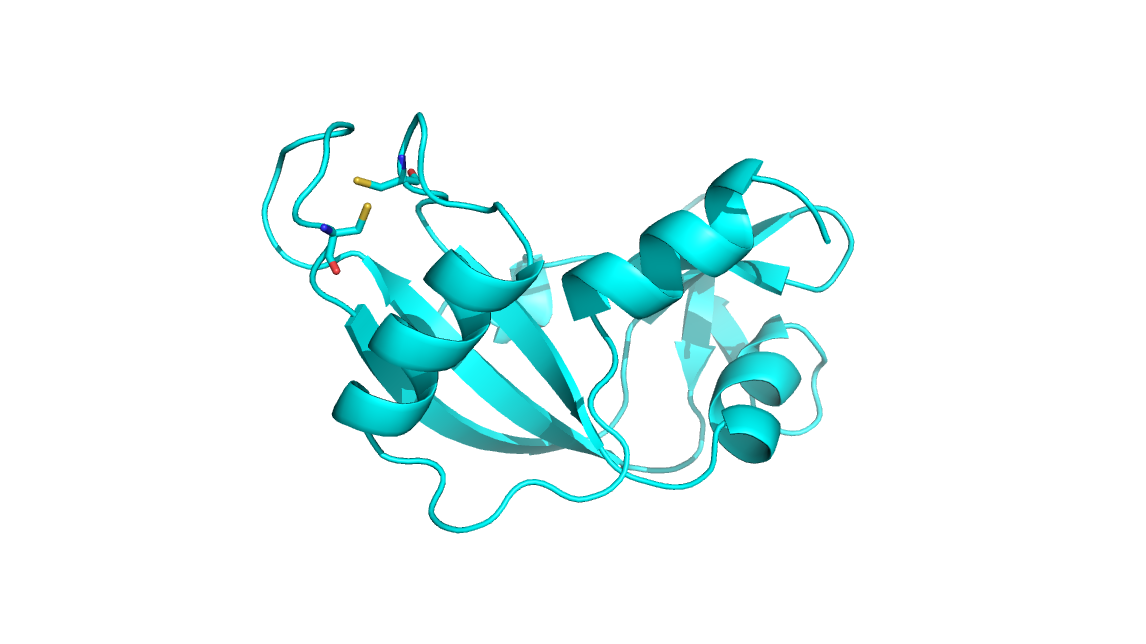
file shows, fig* tleap.png*) . The minimized ambpdb prepared .pdb file is
shown in* 222.png*. But I don't know the how to check the connection between
S-S in the minimized structure.
I *want to keep the S-S bond intact during minimization* so that it does not
break. Is there any way for the same.
Hirdesh
On Tue, Sep 28, 2010 at 1:35 PM, Hannes Loeffler <Hannes.Loeffler.stfc.ac.uk
> wrote:
> On Tue, 28 Sep 2010 13:21:45 +0530
> hirdesh kumar <hirdeshs8.gmail.com> wrote:
>
> > Hi,
> > I have done the distance measurement analysis. The distance is always
> > greater than 3 angstrom (suggesting that there is no disulfide
> > bond:2.05 angstrom).
> > Is there any way via which i can make the distance between two atoms
> > rigid for the starting minimization and equilibration.
>
> Have you confirmed that the S-S bond is indeed defined in your topology
> file? With vmd you can try 'vmd -f -parm7 foo.top -crd foo.crd'.
> This will force vmd to show defined bonds rather than from a heuristic
> analysis. If it is defined then you better look into the actual cause
> for the large deviation.
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
Received on Tue Sep 28 2010 - 03:00:03 PDT