Date: Sat, 3 Apr 2010 00:49:06 -0400
Dear Amber users,
I am using Ptraj in Amber 9.0 for RDF ( Radial
distribution function) calculation. My simulation system contains reverse
micelle and I am calculating the site-site radial distribution of organic
molecules ( that form the core of the reverse micelle) around the
surfactant head group ( that form the corona of the reverse micelle).
Given below is the Ptraj input for RDF calculation...
************************************************************
trajin md.crd
center :MOL mass origin
image origin center familiar
center :SUR mass origin
image origin center familiar # here I am wrapping the reverse
micelle into the unit cell ( because I ran system with ' iwrap = 1' )
radial H_O1 0.05 20.0 :SUR.H6 :MOL.O1
radial H_O2 0.05 20.0 :SUR.H6 :MOL.O2
***********************************************************************************************
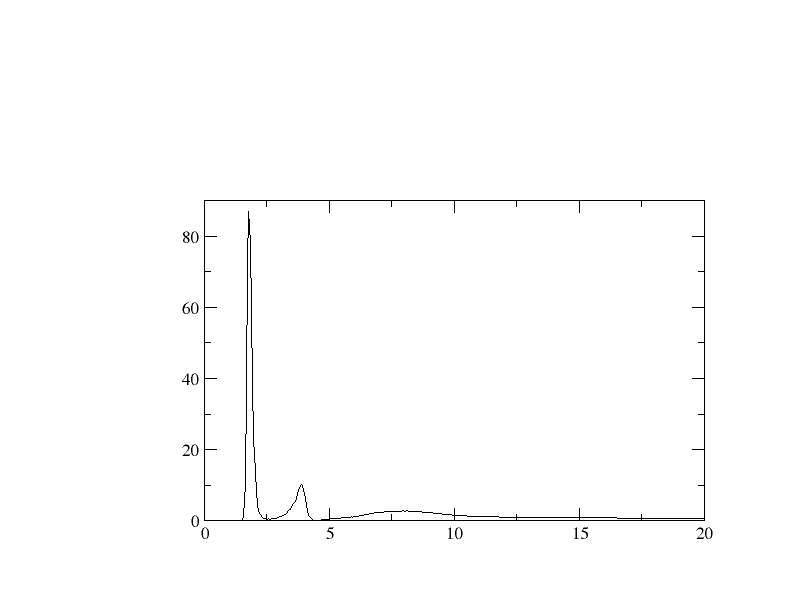
Every thing went fine, but what I am worried about is the maximum value for
g(r) ( maximum value going up to ~80). I browsed through Internet, but
can't able find out any RDF plot with g(r) goes to maximum value like what I
have in my case. As a test run , I calculate water RDF and that went fine.
Am I doing some mistake with calculation? Is it OK with what value I am
getting for g(r)?.
I have attached a RDF plot for your quick reference. Waiting for your
valuable reply
Thanks in advance
Sincerely
Aneesh
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: RDF.png)
