Date: Sat, 4 Jul 2009 13:10:59 +0100
Dear all,
I am trying to use Amber 10 to perform energy minimization and MD on a
system consisting of Guanine tetrads. I have consulted the manual
extensively and followed the online tutorials, but I've still been having
difficulty making the simulations work with periodic boundary conditions. My
main questions are as follows:
1) How would I create a periodic boundary cell with a non-rectangular
lattice?
2) Is it even possible to minimise a system with periodic boundaries (ntb =
1), without the need of an explicit solvent? By 'minimise a system' here I
mean minimise the atoms AND the lattice vectors of the periodic boundary.
3) And finally, would I be able to do NVT molecular dynamics with a system
such as I've just described?
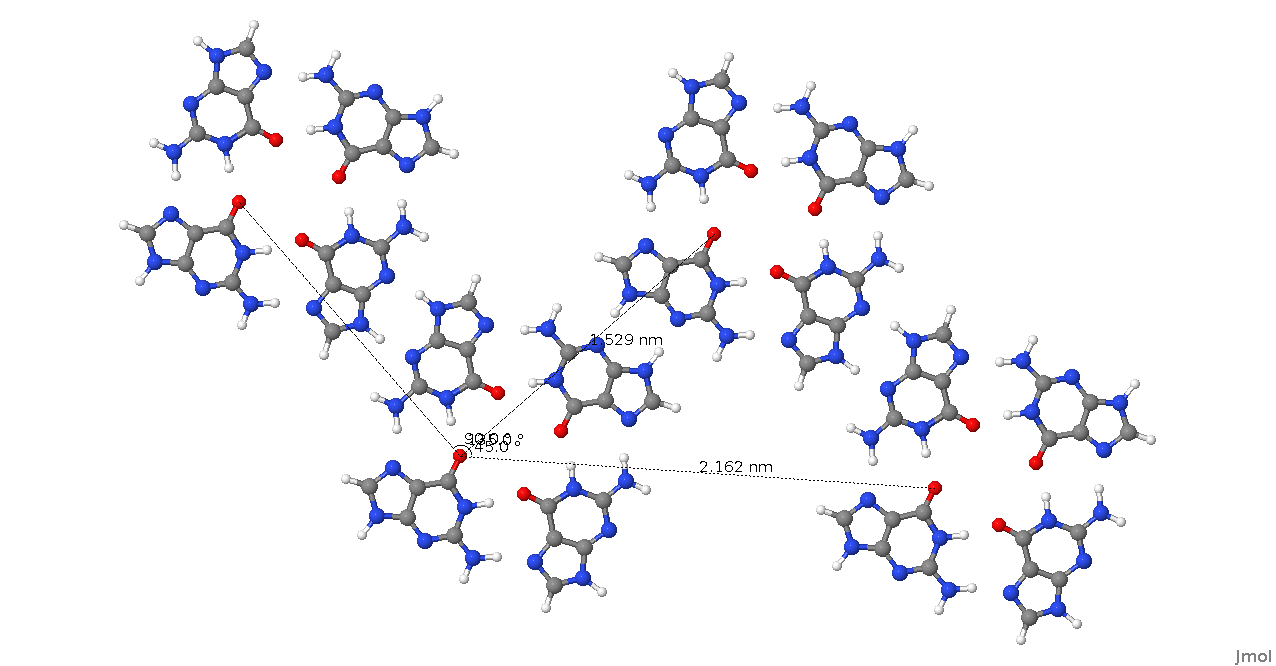
I have attached a picture of a part of the lattice containing 4 Guanine
tetrads with lattice vectors that show what I'm trying to achieve, I'm also
interested in the case where the lattice vectors are not orthogonal. Also
included is the Guanine tetrad .pdb file I've been working with.
Thanks for your time,
Pepe
- chemical/x-pdb attachment: g_tetra.pdb
(image/jpeg attachment: Tetra_lattice.jpg)
- text/plain attachment: ATT00002.txt
