Date: Tue, 4 Sep 2007 09:31:25 -0500
Dear Amber Community,
I am running decomposition energy calculations for one protein-ligand
complex using mm_pbsa. The protein has 389 residues. The run went on fine.
Total GBTOTAL is same as that from binding energy calculation.
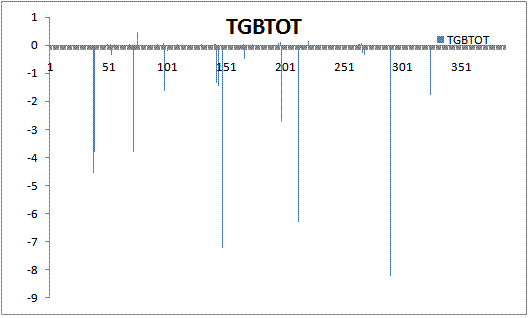
>From the data, it seems that the last residue no. 389 is contributing the
most (-34.8) in the total energy (-78.8), although this residue is far away
from active site. I am confused, how one residue can contribute almost 40%
of the binding energy, even if it is not a part of active site. I have
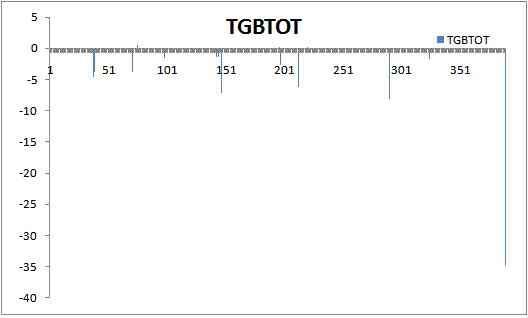
plotted the delta_energy results against residue number. I am attaching the
two plots, one with last residue included and the other with the last one
omitted. I am also attaching the final output file from DC run
"snapshot_statistics.out".
Could you please tell me, if I am missing something in the data
interpretation?
Thanks and Regards
Pankaj
===================================================================
If your ship doesn't come in, swim out to it.......Jonathan Winters
===================================================================
Pankaj R. Daga |
Department of Medicinal Chemistry | e-mail: pdaga_at_olemiss.edu
417 Faser Hall, School of Pharmacy | fax: +1-662-915-5638
University of Mississippi | phone: +1-662-915-1853
University, MS, 38677-1848 |
===================================================================
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo.scripps.edu
- application/octet-stream attachment: snapshot_statistics.out
(image/gif attachment: chart1.GIF)
(image/gif attachment: chart2.GIF)
