Date: Thu, 30 Jun 2005 01:22:04 +0900
Dear D. Case,
Thanks a lot for your time.
I am sorry for making time to understand the things. However, I need to
learn it with some more help from you.
>To get relative free energies of binding, you would need to convert
>ALA->GLY
>in the presence of the ligand, and in its absence. This would involve
>changing both the charges and the vdW parameters.
I got confused. My thermodynamics cycle will look like below. P1 = Protein
with Ala and P2 = protein with GLY , D=drug
G1
P1(sol)+D(sol) ---------> P1-D(sol)
|| ||
G3 || || G4
|| ||
P2(sol)+D(sol) ---------> P2-D(sol)
G2
Thereofe G1-G2 = G3-G4, D(sol) energies will be cancelled out in the
calculation. This essentially means, as you have suggested, I need two TI
calculations for ALA--> GLY with and without the drug molecule.
>> This would involve changing both the charges and the vdW parameters.
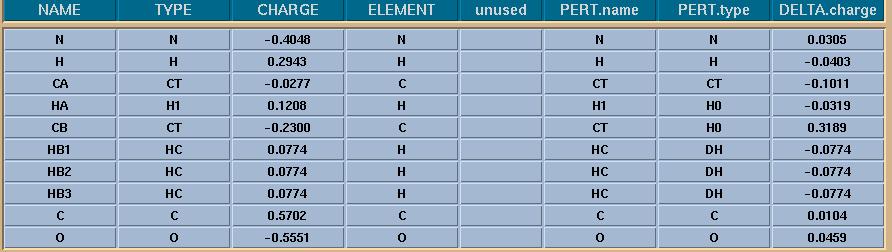
Therefore, both the charge and vdW parameters changes are to be done in one
step with the changes as in the tmp5.jpg file and with the frcmod.DH file as
below. [I am sorry this is the part I confuse most]
ALA to GLY
MASS
DH 1.008
BOND
H0-DH 340.0 1.090 !CT-HC
ANGLE
DH - H0 - DH 35.0 109.50 !HC-CT-HC
CT - H0 - DH 35.0 109.50 !HC-CT-HC
DIHE
- The default values as given by leap
-- -- NONB DH 1.000 0.00 !is it right ? Am I still doing something very stupid? Sincerely, Jiten ----- Original Message ----- From: "David A. Case" <case.scripps.edu> To: <amber.scripps.edu> Sent: Wednesday, June 29, 2005 11:30 PM Subject: Re: AMBER: Re: TI tutorial > On Wed, Jun 29, 2005, Jiten wrote: >> >> 1. For the changing in the charges - I make the DELTA.charge column such >> that the charges of the perturbed state is for GLY. (tmp3.tif is the >> xleap >> editor image) Now do the TI calculations in gas and solvent phase. Find >> the >> difference. Do I need to carry out 12 windows in both the cases ? > > This discussion is not really being very productive I think. I don't > understand why you want to do gas-phase calculations in the first place. > You should think carefully about the thermodynamic cycle you are trying to > apply. > > "DELTA.charge" is the difference between the charge in the lambda = 0 > state > and the lambda=1 state. If you have concerns, study the perturbed prmtop > file to make sure it is getting what you want. Run (short) > simulations with lambda=1, and compare the results to a "standard" prmtop > file that represents the lambda=1 (GLY) state. > > ....dac > > ----------------------------------------------------------------------- > The AMBER Mail Reflector > To post, send mail to amber.scripps.edu > To unsubscribe, send "unsubscribe amber" to majordomo.scripps.edu > > >
-----------------------------------------------------------------------
The AMBER Mail Reflector
To post, send mail to amber.scripps.edu
To unsubscribe, send "unsubscribe amber" to majordomo.scripps.edu
(image/jpeg attachment: tmp5.jpg)
