Date: Thu, 26 Jun 2025 14:09:29 -0400
Dr Anselm,
Thank you for the suggestion. As per your suggestion, I resolved the charge
symmetry issue using the following script.
antechamber -i ligand.mol2 -fi mol2 -o UNL.mol2 -fo mol2 -c abcg2 -s 2 -pf
y -at gaff2 -nc 0 -j 4
However, I noticed something in the resulting MOL2 (UNL.mol2 is attached)
file: the bond order. The MOL2 file generated by Antechamber assigns a
double bond only to C1=N.
While this structure is not wrong, the most probable resonance structure
for my ligand would involve a double bond between C1 and either N1 or
N2—the terminal nitrogens. The same applies to the terminal oxygens:
ideally, the bond between C11 and either O12 or O13 should be a double bond.
Should I leave the MOL2 file as it is (even though the bond information may
not be chemically ideal), or should I manually adjust the bond orders
before proceeding with the simulations? Specifically, should I modify the
structure to reflect either C1–N (single), C1=N1 (double), or C1–N2, and
similarly for the oxygen as C11=O12 or C11–O13?
Thanks!
*Pitambar Poudel*
Graduate Research Assistant
Computational Biophysics and Bioinformatics Lab
Department of Physics and Astronomy, Clemson University
*Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/lab/>*
On Tue, Jun 24, 2025 at 6:35 AM Pitambar Poudel <pitambp.g.clemson.edu>
wrote:
> Hello Anslem,
>
> Thanks for the clarification.
>
> The experimental structure is not available. The structure for protein was
> found via homology modeling and then I used Autodock to dock the ligand
> into the binding pocket. I opend the docked complex in Chimera and saved
> the mol2 of ligand from chimera and used antechamber to generate a new
> mol2, prep, lib, frcmod files.
>
>
> *Pitambar Poudel*
> Graduate Research Assistant
> Computational Biophysics and Bioinformatics Lab
> Department of Physics and Astronomy, Clemson University
> *Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/lab/>*
>
>
> On Tue, Jun 24, 2025 at 6:16 AM Dr. Anselm Horn via AMBER <
> amber.ambermd.org> wrote:
>
>> Pitambar,
>>
>> the resonance in your structure is not reflected in the BOND section of
>> the mol2 file, but is taken into account by the choice of atom types.
>>
>> At a first glance, I do not see anything obvious strange in your mol2
>> file, apart from the large difference in atomic charge of the two
>> guanidinium nitrogen atoms, -0.5978 and -1.0190.
>> In standard Amber force fields, the two nitrogen atoms of the
>> guanidinium group in Arginine have the same (symmetrized) atomic charge.
>>
>> But the situation is not that straightforward:
>> For the free ligand in solution a description with symmetrized charges
>> could be sufficient. When bound to a protein target, however,
>> polarization takes place that might not be described well by fixed
>> atomic charges stemming from the isolated structure.
>>
>> Since you found that the ligand dissociates from the protein, "wrong"
>> atomic charges could be the reason. Have a look at the binding pocket
>> and the protein's ligand interaction there. Maybe simply using
>> symmetrized nitrogen charges solves your problem (if your initial
>> structure is an experimentally determined one).
>>
>> Maybe that helps.
>>
>> Best,
>>
>> Anselm
>>
>> Bioinformatik | NHR.FAU
>> Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU)
>> Germany
>>
>>
>>
>> Am 23.06.2025 um 19:48 schrieb Pitambar Poudel:
>> > Thank you very much for the reply,
>> >
>> > As suggested, I'm currently reviewing the parameter files for a ligand
>> > that were generated by Antechamber. One thing I'm confused about is how
>> > AMBER or Antechamber handles resonance, especially in cases involving
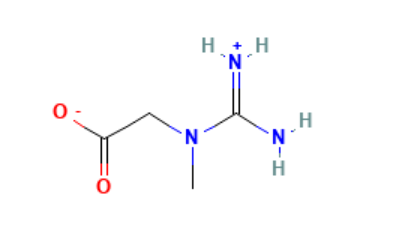
>> > delocalized double bonds. For example, my ligand is a zwitterion when
>> > bound to the protein. One end contains a carboxylate group (COO⁻), and
>> > the other end has two NH₂⁺ groups attached to a single carbon, forming a
>> > guanidinium-like structure. The double bond between the central carbon
>> > and one of the nitrogen atoms can resonate between the two nitrogens,
>> > just like the two C–O bonds in the carboxylate group are also
>> > delocalized. I have attached the image of the structure and the mol2
>> > file created.
>> >
>> > My question is: how is this resonance represented in the MOL2 file? Does
>> > it specify one C=N and one C–N, or both as C=N C=N? And for the
>> > carboxylate group, is it written as one C=O and one C–O⁻ or C=O for
>> > both, or is the resonance captured differently? An incorrect or
>> > incomplete representation of these resonance structures be the reason
>> > why the ligand flies away during molecular dynamics simulations since
>> > this double bond is very important to maintain the zwitterion form
>> >
>> > *Pitambar Poudel*
>> > Graduate Research Assistant
>> > Computational Biophysics and Bioinformatics Lab
>> > Department of Physics and Astronomy, Clemson University
>> > /Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/lab/>/
>> >
>> >
>> > On Fri, Jun 20, 2025 at 5:42 AM Dr. Anselm Horn via AMBER
>> > <amber.ambermd.org <mailto:amber.ambermd.org>> wrote:
>> >
>> > Pitambar,
>> >
>> > your guess about a ligand parameterization issue seems reasonable
>> to me,
>> > if you start from a known complex structure.
>> > I'd suggest to check the ligand parameters, i.e. atom types and
>> charges,
>> > as well as the structural elements (planarity vs. non-planarity) of
>> the
>> > ligand (=> minimization/simulation of the free ligand).
>> > Additionally, ensure that the ligand has the correct molecular
>> charge
>> > and protonation state when simulating the bound state.
>> > Maybe key polar interactions between ligand and protein are not
>> > correctly modelled and you want to try a different charge generation
>> > method.
>> >
>> > If you do not start from a known complex structure, then the protein
>> > environment of the binding pocket might disfavor ligand binding:
>> your
>> > protein could have different conformations in bound and unbound
>> state.
>> >
>> > Maybe that helps.
>> >
>> > Best,
>> >
>> > Anselm
>> >
>> > Bioinformatik | NHR.FAU
>> > Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU)
>> > Germany
>> >
>> >
>> > Am 19.06.2025 um 22:57 schrieb Pitambar Poudel via AMBER:
>> > > Hello all,
>> > > I’m working on a system consisting of a transporter protein with a
>> > ligand
>> > > positioned at a binding pocket and the whole system embedded in a
>> > lipid
>> > > bilayer. The bilipid layer was constructed using packmol-memgen.
>> > Before
>> > > that, I used Antechamber for ligand parameterization with the
>> > following
>> > > script:
>> > >
>> > >
>> > > *antechamber -i ligand.mol2 -fi mol2 -o UNL.mol2 -fo mol2 -c abcg2
>> > -s 2 -pf
>> > > y -j 5 -at gaff2 -nc 0 antechamber -i UNL.mol2 -fi mol2 -o
>> > UNL.prep -fo
>> > > prepi -c abcg2 -s 2 -pf y -j 5 -at gaff2 -nc 0 parmchk2 -i
>> UNL.prep -f
>> > > prepi -o UNL.frcmod -s 2*
>> > > During equilibration, with gradually decreasing positional
>> > restraints (~100
>> > > ns), the system remains stable. However, once restraints are fully
>> > lifted
>> > > in the production run, the ligand instantly dissociates and flies
>> > away.
>> > > Initially, I had used -j 4, but some double bonds were incorrectly
>> > assigned
>> > > during parametrization, so I switched to -j 5 as it allows to
>> read the
>> > > connectivity table from the input and then run ’bondtype’ and
>> > ’atomtype’
>> > > sequentially. My current guess is that the issue lies in ligand
>> > > parametrization—possibly incorrect charges or missing parameters.
>> > I don't
>> > > see issues after running parmchk2, however. Any suggestions or
>> > insights
>> > > would be appreciated.
>> > > *Pitambar Poudel*
>> > > Graduate Research Assistant
>> > > Computational Biophysics and Bioinformatics Lab
>> > > Department of Physics and Astronomy, Clemson University
>> > > *Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/>
>> > <http://compbio.clemson.edu/lab/>*
>> > > _______________________________________________
>> > > AMBER mailing list
>> > > AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>> > > http://lists.ambermd.org/mailman/listinfo/amber
>> > >
>> >
>> >
>> > _______________________________________________
>> > AMBER mailing list
>> > AMBER.ambermd.org <mailto:AMBER.ambermd.org>
>> > http://lists.ambermd.org/mailman/listinfo/amber
>> >
>>
>>
>> _______________________________________________
>> AMBER mailing list
>> AMBER.ambermd.org
>> http://lists.ambermd.org/mailman/listinfo/amber
>>
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- chemical/x-mol2 attachment: UNL.mol2
(image/png attachment: structure.png)
