Date: Mon, 23 Jun 2025 13:48:34 -0400
Thank you very much for the reply,
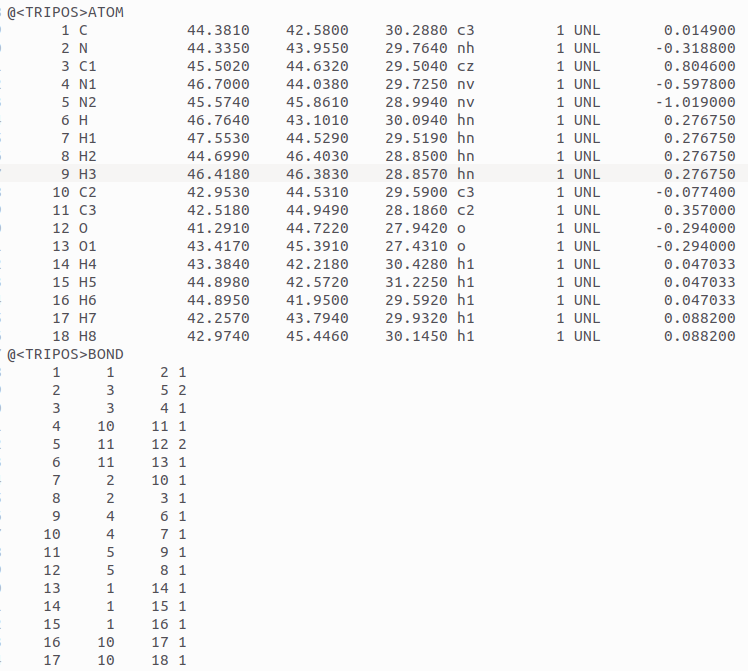
As suggested, I'm currently reviewing the parameter files for a ligand that
were generated by Antechamber. One thing I'm confused about is how AMBER or
Antechamber handles resonance, especially in cases involving delocalized
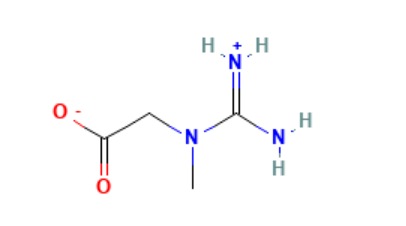
double bonds. For example, my ligand is a zwitterion when bound to the
protein. One end contains a carboxylate group (COO⁻), and the other end has
two NH₂⁺ groups attached to a single carbon, forming a guanidinium-like
structure. The double bond between the central carbon and one of the
nitrogen atoms can resonate between the two nitrogens, just like the two
C–O bonds in the carboxylate group are also delocalized. I have attached
the image of the structure and the mol2 file created.
My question is: how is this resonance represented in the MOL2 file? Does it
specify one C=N and one C–N, or both as C=N C=N? And for the carboxylate
group, is it written as one C=O and one C–O⁻ or C=O for both, or is the
resonance captured differently? An incorrect or incomplete representation
of these resonance structures be the reason why the ligand flies away
during molecular dynamics simulations since this double bond is very
important to maintain the zwitterion form
*Pitambar Poudel*
Graduate Research Assistant
Computational Biophysics and Bioinformatics Lab
Department of Physics and Astronomy, Clemson University
*Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/lab/>*
On Fri, Jun 20, 2025 at 5:42 AM Dr. Anselm Horn via AMBER <amber.ambermd.org>
wrote:
> Pitambar,
>
> your guess about a ligand parameterization issue seems reasonable to me,
> if you start from a known complex structure.
> I'd suggest to check the ligand parameters, i.e. atom types and charges,
> as well as the structural elements (planarity vs. non-planarity) of the
> ligand (=> minimization/simulation of the free ligand).
> Additionally, ensure that the ligand has the correct molecular charge
> and protonation state when simulating the bound state.
> Maybe key polar interactions between ligand and protein are not
> correctly modelled and you want to try a different charge generation
> method.
>
> If you do not start from a known complex structure, then the protein
> environment of the binding pocket might disfavor ligand binding: your
> protein could have different conformations in bound and unbound state.
>
> Maybe that helps.
>
> Best,
>
> Anselm
>
> Bioinformatik | NHR.FAU
> Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU)
> Germany
>
>
> Am 19.06.2025 um 22:57 schrieb Pitambar Poudel via AMBER:
> > Hello all,
> > I’m working on a system consisting of a transporter protein with a ligand
> > positioned at a binding pocket and the whole system embedded in a lipid
> > bilayer. The bilipid layer was constructed using packmol-memgen. Before
> > that, I used Antechamber for ligand parameterization with the following
> > script:
> >
> >
> > *antechamber -i ligand.mol2 -fi mol2 -o UNL.mol2 -fo mol2 -c abcg2 -s 2
> -pf
> > y -j 5 -at gaff2 -nc 0 antechamber -i UNL.mol2 -fi mol2 -o UNL.prep -fo
> > prepi -c abcg2 -s 2 -pf y -j 5 -at gaff2 -nc 0 parmchk2 -i UNL.prep -f
> > prepi -o UNL.frcmod -s 2*
> > During equilibration, with gradually decreasing positional restraints
> (~100
> > ns), the system remains stable. However, once restraints are fully lifted
> > in the production run, the ligand instantly dissociates and flies away.
> > Initially, I had used -j 4, but some double bonds were incorrectly
> assigned
> > during parametrization, so I switched to -j 5 as it allows to read the
> > connectivity table from the input and then run ’bondtype’ and ’atomtype’
> > sequentially. My current guess is that the issue lies in ligand
> > parametrization—possibly incorrect charges or missing parameters. I don't
> > see issues after running parmchk2, however. Any suggestions or insights
> > would be appreciated.
> > *Pitambar Poudel*
> > Graduate Research Assistant
> > Computational Biophysics and Bioinformatics Lab
> > Department of Physics and Astronomy, Clemson University
> > *Lab: http:/compbio.clemson.edu/ <http://compbio.clemson.edu/lab/>*
> > _______________________________________________
> > AMBER mailing list
> > AMBER.ambermd.org
> > http://lists.ambermd.org/mailman/listinfo/amber
> >
>
>
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
>
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: mol2.png)
(image/png attachment: structure.png)
