Date: Tue, 4 Mar 2025 10:16:41 +0000
Hi, I encountered a problem when parametrizing a ligand containing a 5,5-fused aromatic system, specifically imidazo[2,1-b][1,3,4]thiadiazole. Using the input structure obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/compound/88013-15-4), I executed the following commands. ``` antechamber -i ITD.sdf -fi sdf -o ITD_abcg2.mol2 -fo mol2 -c abcg2 -at gaff2 -nc 0 -rn LIG parmchk2 -i ITD_abcg2.mol2 -o ITD_abcg2.frcmod -f mol2 -s gaff2 ``` Then I executed tleap to obtain the prmtop and inpcrd files. ``` source leaprc.gaff2 source leaprc.water.tip3p lig = loadMol2 ITD_abcg2.mol2 loadAmberParams ITD_abcg2.frcmod solvateBox lig TIP3PBOX 10.0 saveAmberParams lig lig.prmtop lig.inpcrd quit ``` After energy minimization, I got a strange structure in which the fused system bended at the bridgehead position (see minimized.png). At first, I thought this was caused by IMPROPER parameters written in the frcmod file. But the parameters seem OK as the default values for the benzene ring was assigned for the bridge head positions (see lig.frcmod and atomtype.png). How can I solve this issue? I would appreciate your help. Best regards, Taiyo Yokoi
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
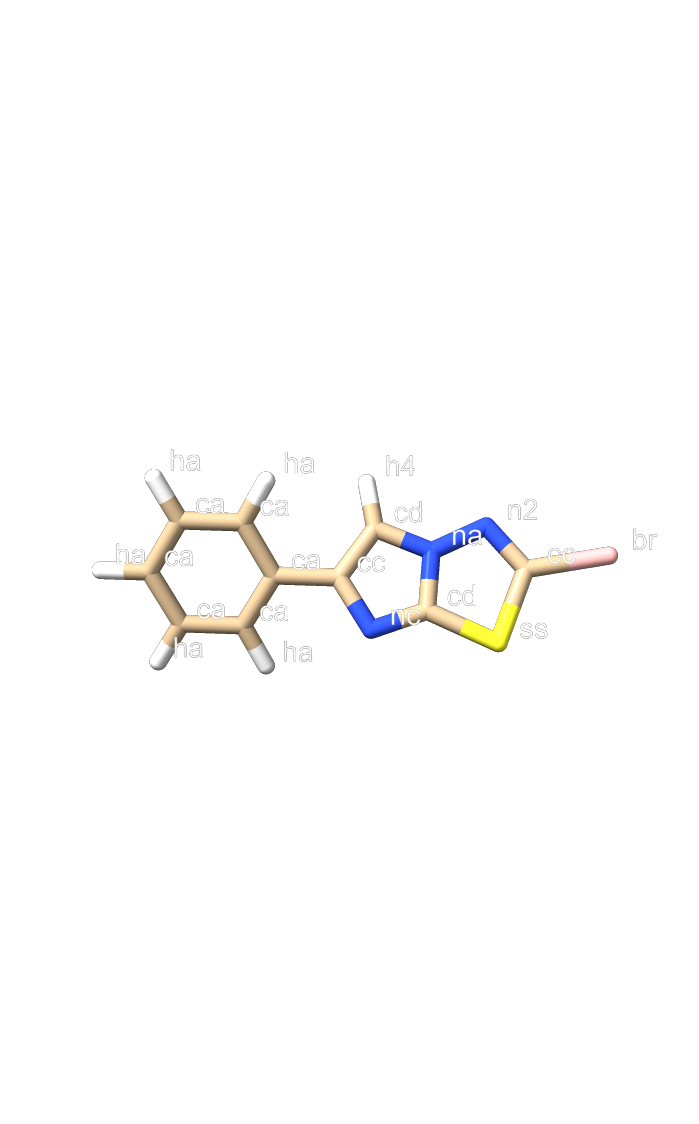
(image/png attachment: atomtype.png)
- application/octet-stream attachment: ITD_abcg2.frcmod
- chemical/x-mol2 attachment: ITD_abcg2.mol2
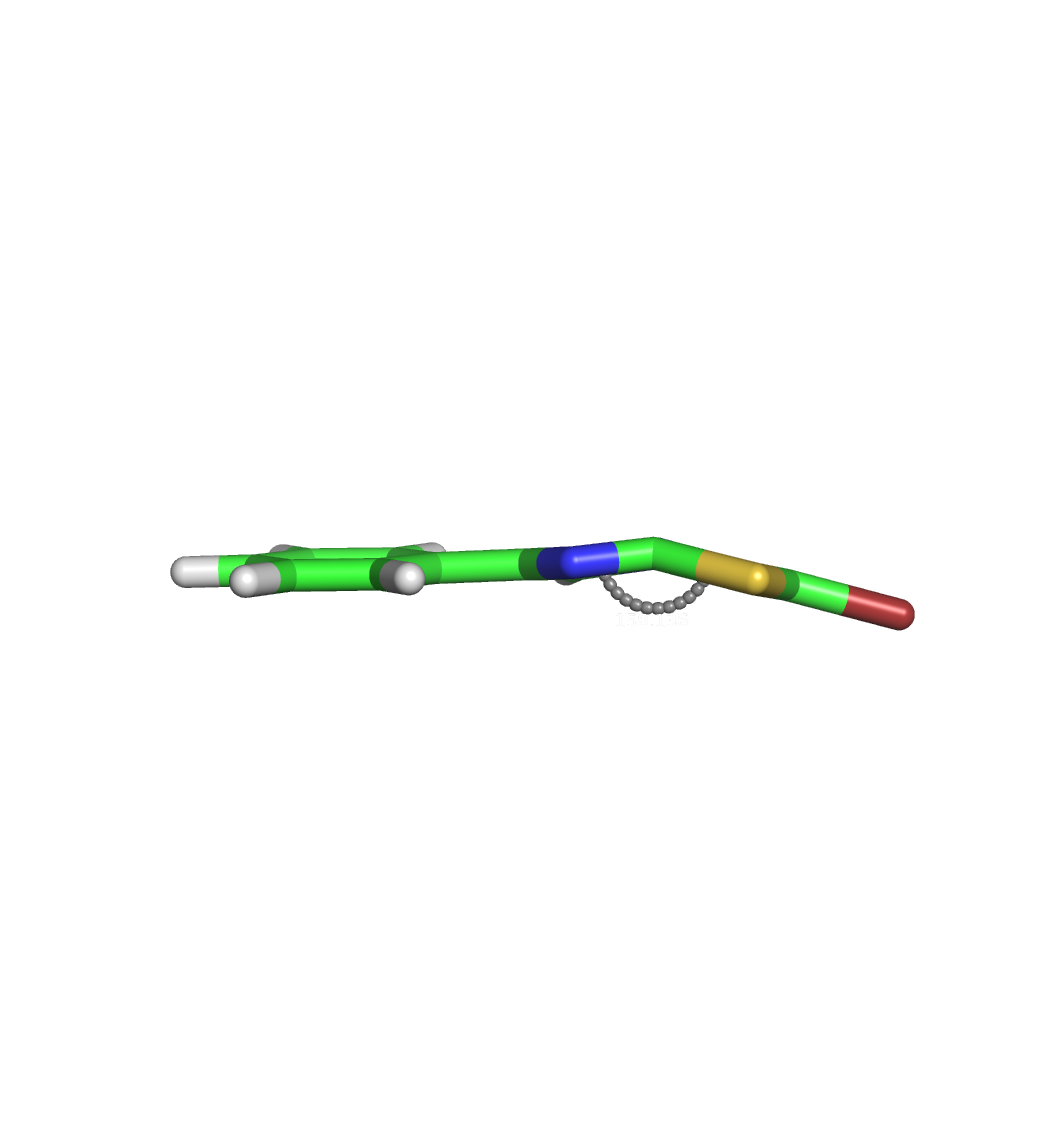
(image/png attachment: minimized.png)
