Date: Sun, 5 Jan 2025 18:09:03 +0530
Dear Experts,
I am trying to run aMD simulation using amber24. I set the following
parameters in the mdin file
iamd=1,
ethreshp=-159511.27,
alphap=8490.88,
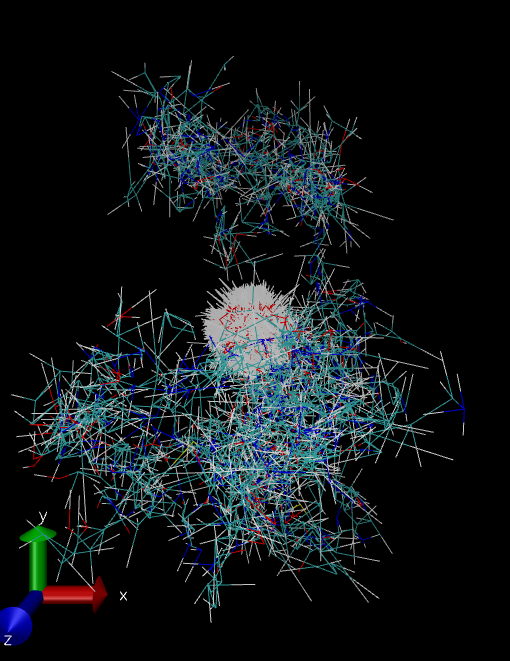
aMD simulation runs properly. But when I visualize the trajectory I see the
following snapshotr of the protein.
Is this correct? If not, what can I do? How do I set the aMD parameter?
-- *With regards,* *Dulal Mondal,* *Research Scholar,* *Department of Chemistry,* *IIT Kharagpur, Kharagpur 721302.*
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Screenshot_from_2025-01-05_18-07-05.png)
