Date: Tue, 31 Dec 2024 18:02:50 +0000
Hi Amber team,
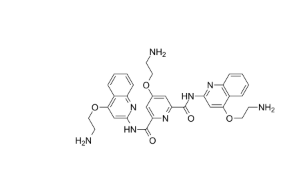
Seems like I am making a mistake in my antechamber protocol for this small molecule (PDS). It should not be so complicated. All I have done is docked it and saved the docked pose as a sdf file which I then converted to pdb.
[cid:4452caa5-3ec9-4c98-b12a-87ddb7d1fbc8]
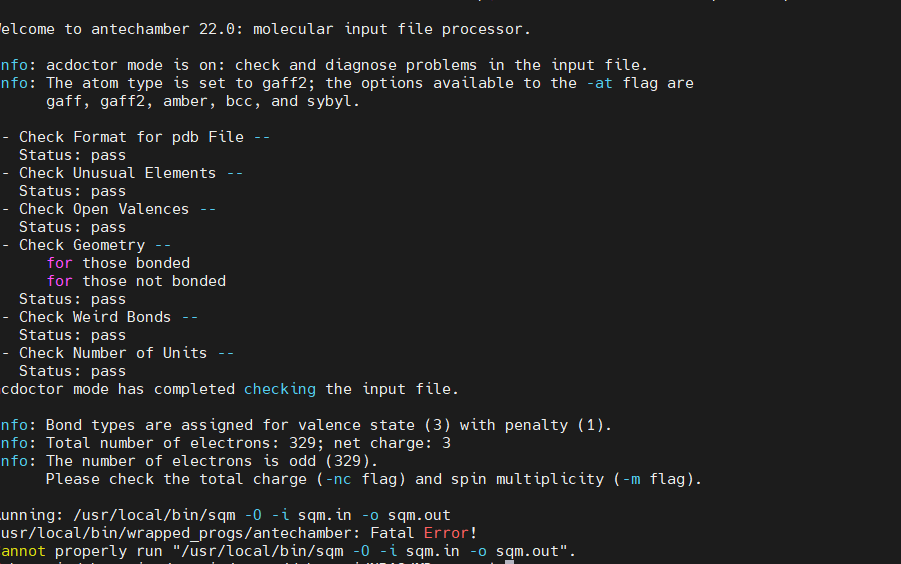
It has 3 basic amines and I would anticipate the net charge is 3 but when I run the antechamber command I get this error:
[cid:1111194d-e767-4766-a294-cfc8db89ec62]
--------------------------------------------------------
AMBER SQM VERSION 19
By
Ross C. Walker, Michael F. Crowley, Scott Brozell,
Tim Giese, Andreas W. Goetz,
Tai-Sung Lee and David A. Case
--------------------------------------------------------
--------------------------------------------------------------------------------
QM CALCULATION INFO
--------------------------------------------------------------------------------
QMMM: System specified with odd number of electrons ( 237)
QMMM: but odd spin ( 1). You most likely have the charge of
QMMM: QM region (qmcharge) set incorrectly. Correct error and re-run calculation.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
(image/png attachment: 02-image.png)
- application/octet-stream attachment: PDS_1.sdf
