Date: Tue, 10 Dec 2024 15:32:33 +0100
Le 09/12/2024 à 14:08, Daniel Roe via AMBER a écrit :
> Hi,
>
> Note that as of version 6.29.9 (currently on GitHub) there is a new
> experimental keyword for ‘autoimage’ that should work much better for
> membrane systems:
>
> autoimage mode byvec
>
> If you get a chance to try it please let me know how it worked for you!
>
> -Dan
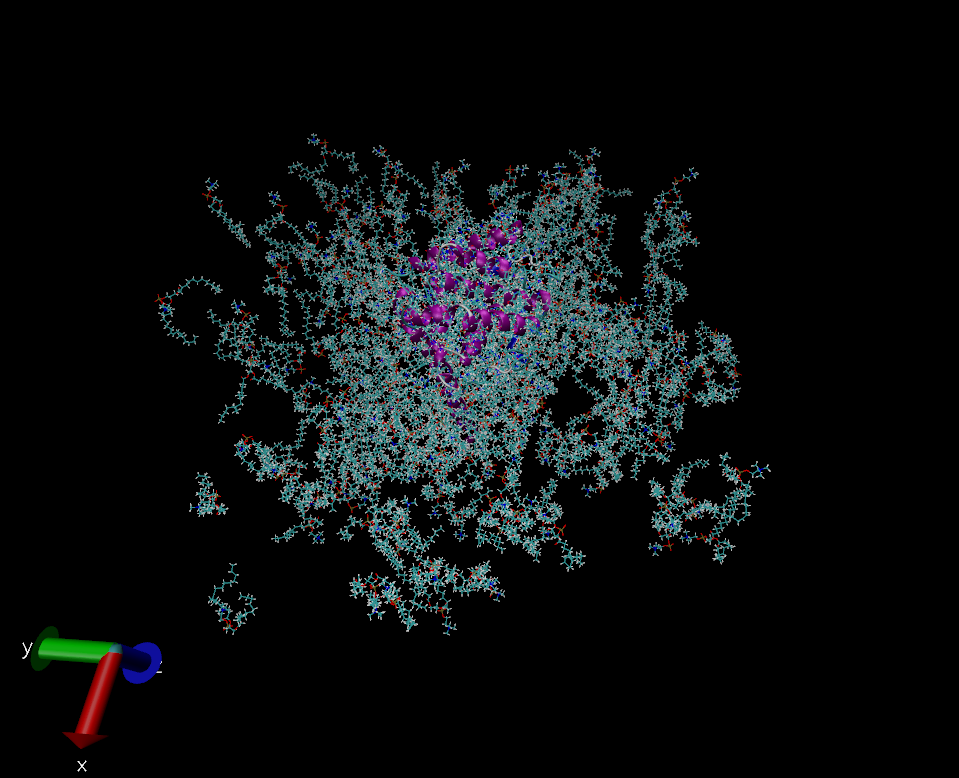
Tried the latest version available (your commit from December 6th), and :
(a) good news, the protein is correctly packed AND at the center of the
lipids
(b) good news, the lipids do not jump from one box to the other like
they do on autoimage alone
(c) bad news, the lipids are in a much bigger box (xy plane is fine), so
the membrane seems porous (see screenshot).
I can share off the list the files and more details if you wish to have
a benchmark.
Best,
Stéphane
-- Maître de conférences Hors Classe, PEDR, HDR US2B, Nantes Université, CNRS, UMR 6286, Team Structural Bioinformatics, F-44000 Nantes http://www.us2b.univ-nantes.fr/ - http://www.steletch.org
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: protein_in_porous_membrane_autoimage-mode-byvec.png)
