Date: Fri, 20 Sep 2024 20:33:11 +0000
Dear amber community,
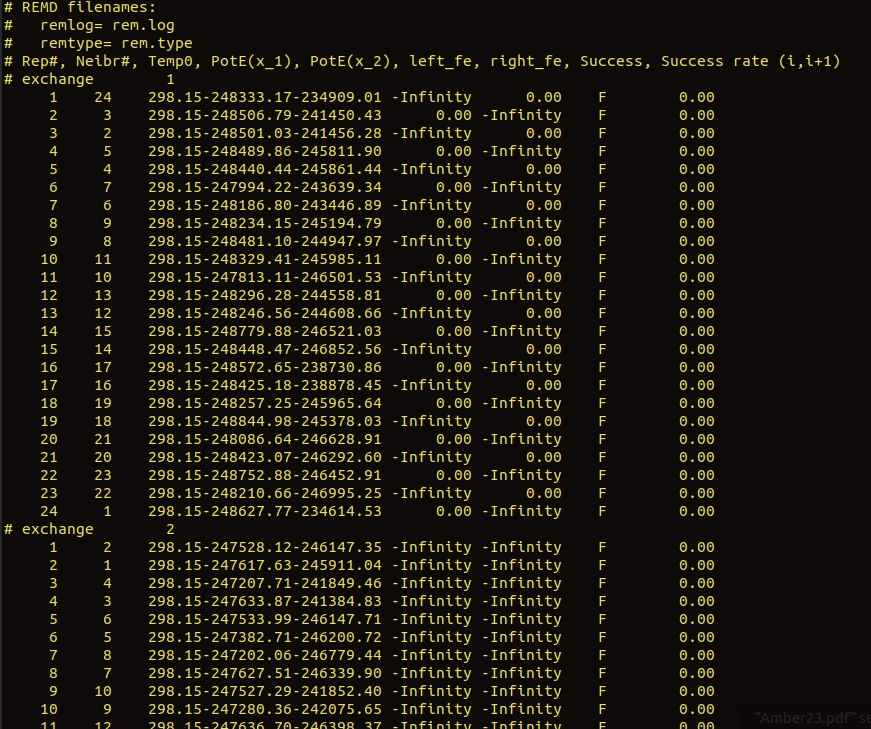
I am trying to run a h-remd for a relatively large system (60404 atoms after solvation). To create the replicas i have changed the distance between CoMs of two residues (as the reaction coordinate) and picked replicas from that. I have conducted the same simulation for a smaller system (setting 0.6 Angstrom reaction coordinate difference between each replica) and did not encounter any issues. But for the larger system I have tried, 0.1,0.2,0.3,0.4 angstrom differences but still not getting any replica exchanges. Infact left_fe, right_fe keep showing '-infinity' (picture attached). Since h-remd is said to be less dependent on the number of replicas (as opposed to t-remd) with the increasing system size, this problem is quite difficult for me to understand. Can anyone give me an idea about this issue or propose a way to calculate the number of replicas for h-remd?
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/jpeg attachment: Selection_020.jpg)
