Date: Tue, 6 Aug 2024 22:06:50 +0530
Hi Amy
Thanks for the email.
I apologise for any confusion
caused by the term 'chunks'.
To clarify, I was NOT referring to
the various interval-based
averages namely, cumulative,
running, and chunk, that can be
obtained from cphstats.
I meant 'chunk' in the context of
Jason Swail's tutorial on pH-REMD:
http://jswails.wikidot.com/ph-remd#toc10
To quote from the tutorial:
> If you have run 10 ns of simulation
in 1 ns chunks, you will need to
run this program 10 times!
>
PFA two titration curves: one obtained from
by processing just the first segment, the other
obtained by processing concatenated cpouts
from all segments.
Thanks and regards
Nitin Kulhar
On Mon, Aug 5, 2024 at 4:35 AM He, Amy <he.1768.buckeyemail.osu.edu> wrote:
> Hi Nitin,
>
>
>
> Why do you think the results are wrong? Did it cumulate, or not cumulate,
> or not space in the way you want?
>
> The manual will give you a detailed difference with examples for the
> Cumulative, running, and “chunk” options of cphstats.
>
>
>
>
>
> *From: *Nitin Kulhar via AMBER <amber.ambermd.org>
> *Date: *Friday, August 2, 2024 at 9:38 PM
> *To: *AMBER Mailing List <amber.ambermd.org>
> *Subject: *[AMBER] cphstats processing of pH-REMD 'chunks'
>
>
>
> Dear all
>
> I performed pH-REMD of 8
> REPLICAS. 25 ns of REMD
> were run in 1 ns SEGMENTS
> (or chunks) as delineated in
> the tutorial by Jason Swails
> available at link <
> https://urldefense.com/v3/__http://jswails.wikidot.com/ph-remd__;!!KGKeukY!yNeLxboggV6vrz65BXBmJdJtWYdmVZcrdIOV1wBGc78GZvpfjF_-WCNsdRPGRjBJUhH7F7YBvR3VSKHDhd3UMV574E0$
> >.
>
> *The problem:*
>
> The pKa values obtained by
> processing the cpout files from
> all SEGMENTS (or chunks)
> were questionable, nay, wrong.
> However, the pKa values obtained
> by processing the cpout file from
> the first SEGMENT alone were
> correct.
>
> *Salient details of the REMD:*
>
> The inputs for each SEGMENT
> were generated by the
> genremdinputs.py script.
>
> The cpin and starting coordinates
> for REPLICA 'i' of SEGMENT 'j'
> were respectively the cprestart
> and restart files generated by
> REPLICA 'i' during SEGMENT 'j-1'.
>
> While the seed (ig) value
> differed among REPLICAS
> of the same SEGMENT, it
> was the same for a given
> REPLICA in all SEGMENTS.
> (Is that genremdinputs.py's doing?)
>
> *Salient details of processing cpouts:*
>
> The cpout files from consecutive
> SEGMENTS were concatenated
> with the cat command. This was
> done for each REPLICA separately.
>
> The concatenated cpout files of ALL
> REPLICAS were sorted according to
> pH values by using cphstats program
> with the --fix-remd option.
>
> The pH-sorted cpout files from above
> were processed with cphstats to obtain
> pKa values and population data.
>
> Please help me spot the mistake
> in my logic or execution of the steps.
>
> Thanks and regards
> Nitin Kulhar
>
> --
>
>
> Disclaimer:- This footer text is to convey that this email is sent by one
> of the users of IITH. So, do not mark it as SPAM.
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
>
> https://urldefense.com/v3/__http://lists.ambermd.org/mailman/listinfo/amber__;!!KGKeukY!yNeLxboggV6vrz65BXBmJdJtWYdmVZcrdIOV1wBGc78GZvpfjF_-WCNsdRPGRjBJUhH7F7YBvR3VSKHDhd3UQjpcOUQ$
> <https://urldefense.com/v3/__http:/lists.ambermd.org/mailman/listinfo/amber__;!!KGKeukY!yNeLxboggV6vrz65BXBmJdJtWYdmVZcrdIOV1wBGc78GZvpfjF_-WCNsdRPGRjBJUhH7F7YBvR3VSKHDhd3UQjpcOUQ$>
>
-- Disclaimer:- This footer text is to convey that this email is sent by one of the users of IITH. So, do not mark it as SPAM.
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: fracprot-combined.png)
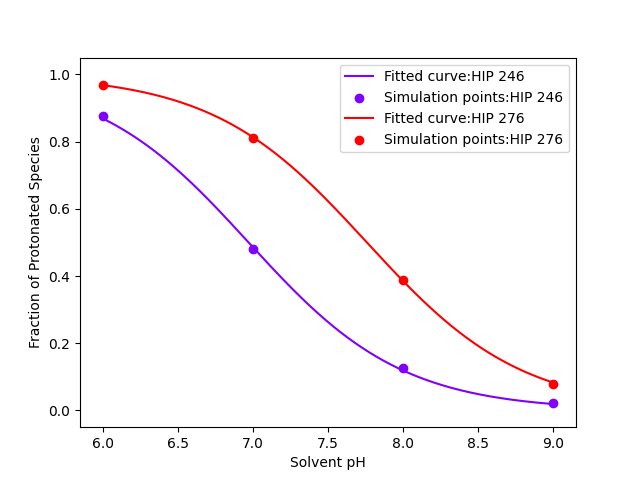
(image/png attachment: fracprot-s-1.png)
