Date: Fri, 17 May 2024 12:13:14 -0400
Hi AMBER community,
I'm having some issues every time I try to add ions to my system that are
different from Na+, K+, and Cl-. For some reason, *frcmod.ionsjc_tip3p* is
not allowing me to use Br- and I-. Here I attached the script that I am
using and the error screen.
Thank you!
-------------------------------------------------------------------------------------------
-------------------------------------------------------------------------------------------
source leaprc.gaff2
source leaprc.water.tip3p
loadamberparams frcmod.mon
loadoff monomer.lib
loadoff atomic_ions.lib
loadamberparams frcmod.ionsjc_tip3p
MOL = loadpdb dimer_old.pdb
#set default nocenter off
#setBox MOL "vdw" 100
#check MOL
addIons2 MOL Na+ 2
addIons2 MOL Br- 2
addIons2 MOL Cl- 0
check MOL
solvatebox MOL TIP3PBOX 30.0
saveamberparm MOL dimer_tip3p_ions.parm7 dimer_tip3p_ions.rst7
savepdb MOL dimer_tip3p_ions.pdb
quit
---------------------------------------------------------------------------------------------
---------------------------------------------------------------------------------------------
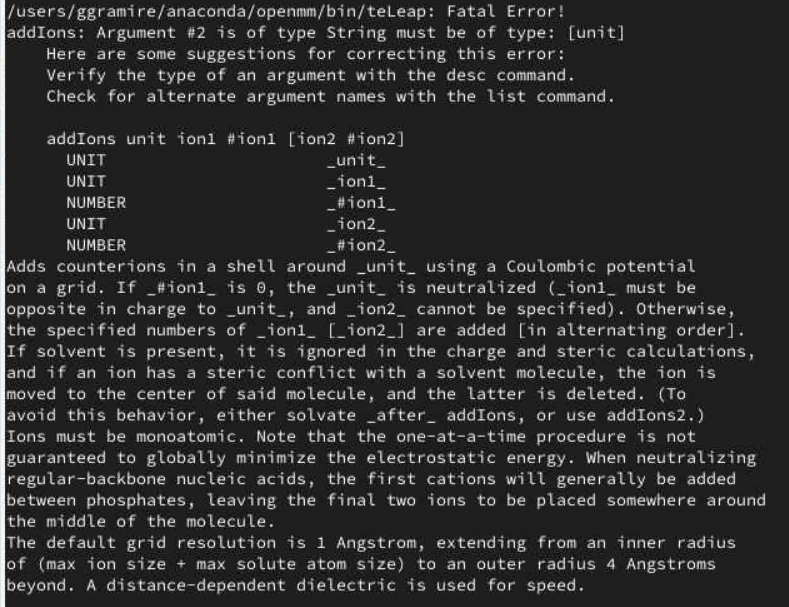
Error screen:
[image: Captura de pantalla (52).png]
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: Captura_de_pantalla__52_.png)
