Date: Thu, 9 May 2024 09:26:32 -0230
Hi All,
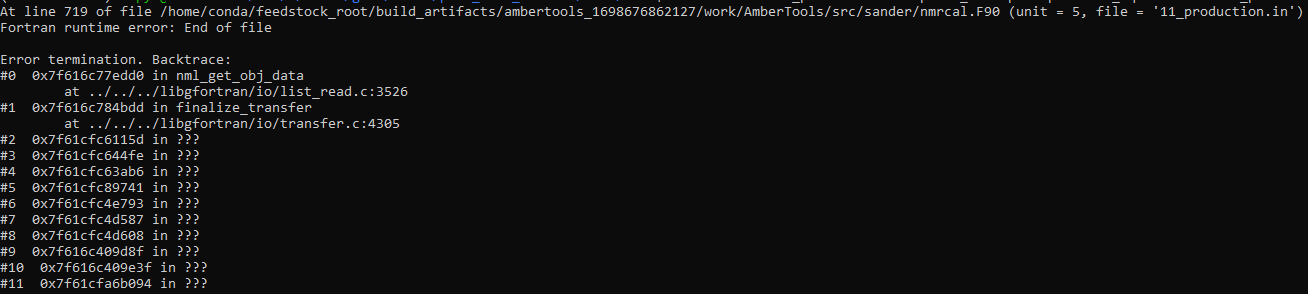
I am trying to run an RNA simulation with an added distance restraint
between two nucleobases using nmropt=1. With nmropt=1 added I get a fortran
end of file runtime error. Without the nmropt and DISANG lines everything
runs fine. What might be causing these errors? Attached are my input file,
DISANG file, and a screenshot of the error message.
Thanks,
Aaron
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- application/octet-stream attachment: 11_production.in
- application/octet-stream attachment: end-distance.restraint
(image/png attachment: error.png)
