Date: Tue, 16 Apr 2024 00:17:24 +0100
Dear AMBER users,
I am using PDB file (code pdb :4w93) structure for molecular dynamics
simulation with AMBER22.
First I ran the command:
- antechamber -i Lig.log -fi gout -o Lig.mol2 -fo mol2 -c resp
- parmchk -i Lig.mol2 -o Lig.frcmod -f mol2
after # xleap
- source leaprc.gaff2
- source leaprc.protein.ff14SB
- source leaprc.water.tip3p
- loadamberparams Lig,frcmod
- MOL=loadmol2 Lig,mol2
- a=loadpdb 4w93,pdb
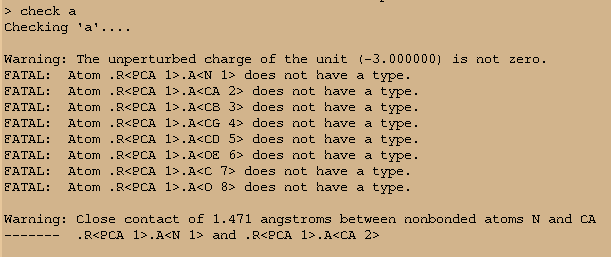
- check a
saveamberparm a Lig.top Lig.crd
quit
But at the last step, the parameters cannot be saved because of this kind
of errors:
[image: image.png]
How can this problem be solved?
Thank you for your help.
Abdelatif
University of Batna
_______________________________________________
AMBER mailing list
AMBER.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
(image/png attachment: image.png)
